How smarter guide RNA design boosted bacterial production of useful chemicals
Published in Bioengineering & Biotechnology and Microbiology
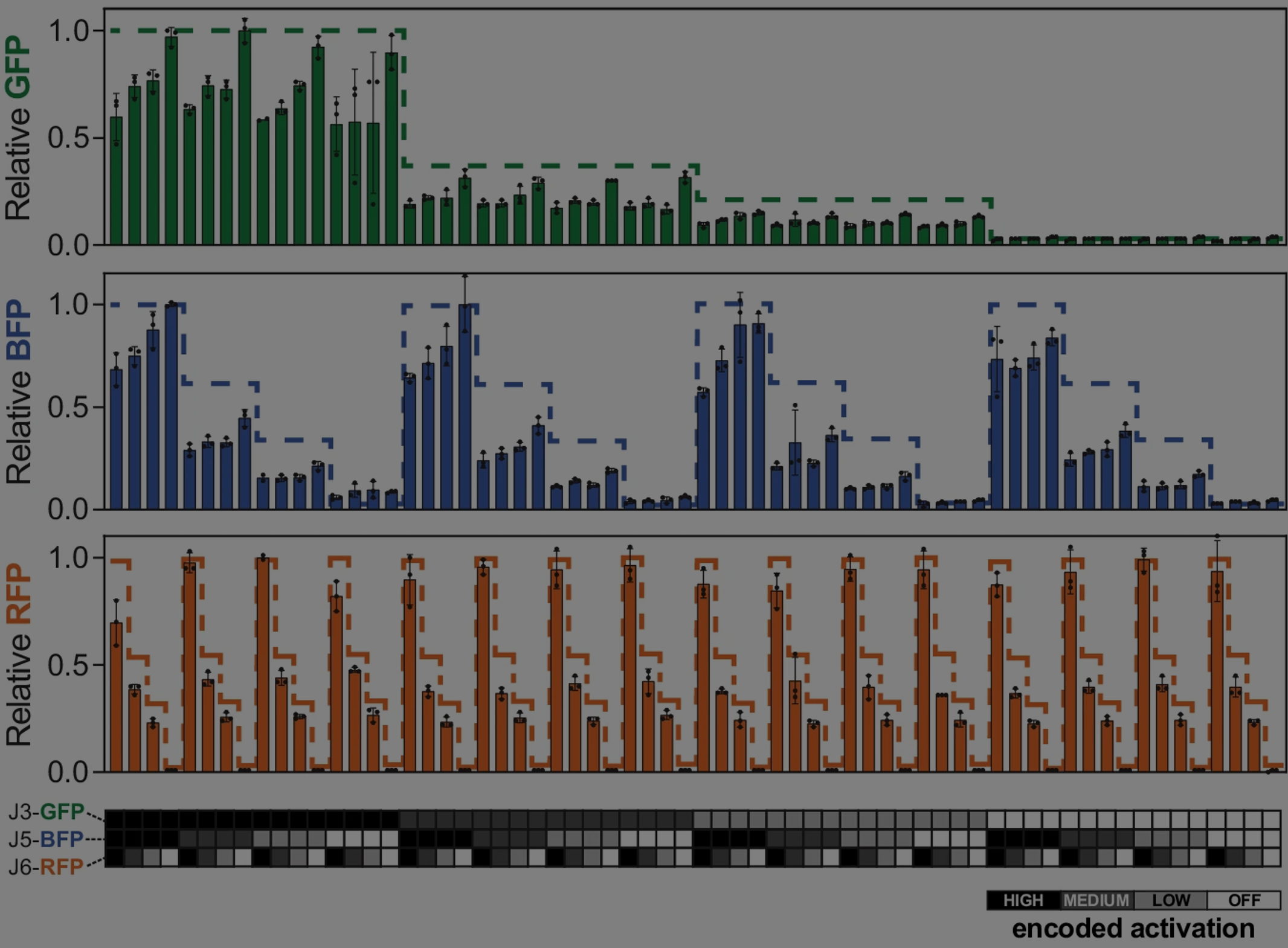
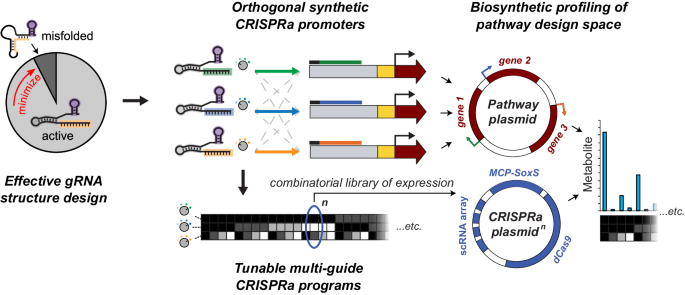
This work started years ago, when I (Ice) had just joined the lab. In 2018, Jason and David were committed to design a highly predictable guide RNA platform by combining computational RNA design with existing CRISPR gene regulation tools we had just developed at the time [1]. The initial results for controlling expression of three fluorescent proteins were beautifully crafted, and that was when I joined the crew to help adapt the technology for metabolic engineering applications. As a synthetic chemist by training, I was exploring the biopterin biosynthetic pathway at that time. Fortunately, this three-gene pathway yields fluorescent small molecules and could be put under control of the three CRISPR activation (CRISPRa) promoters just characterized in the fluorescent protein system. We combinatorially profiled the expression of enzymes in this pathway and found production differences across the design space, as well as some interesting trends. However, this project was paused while our lead members founded a biotech startup using their RNA design expertise. This is not uncommon in the research community, and I want to share our experience on how we brought this project back to life by endeavoring toward metabolic engineering applications.

It’s worth recounting how we got to the point of controlling engineered biosynthetic pathways with CRISPRa. Previously, our team elucidated the design rules and various factors that govern the effectiveness of bacterial CRISPRa in a single channel of expression [2]. If we were to use this tool to program multiple channels simultaneously, as in a multi-enzyme pathway, we needed to ensure that we have comparable levels from each channel and mitigate crosstalk between the channels. Personally, I like to compare this gene regulatory tool to an audio mixer for tuning different volumes of music elements to get the best harmony. We had developed a thorough understanding of CRISPRa promoter design rules [3], but a major challenge remained in the design rules of the guide RNA (gRNA): specifically, how to avoid gRNAs that fold into nonfunctional structures. The specificity and orthogonality of CRISPR systems comes from the 20-nucleotide spacer sequence of the gRNA, but changing this sequence might change how the overall RNA folds, due to interactions between the spacer and the rest of the sequence. A common screening approach in the field is to 1) design a big library of sequences, 2) build and test the physical gRNAs, and then 3) elucidate the design rules later from the acquired results. We used an opposite approach by initially designing gRNAs computationally, before making physical gRNAs, so that they possessed distinct key parameters of RNA folding energetics for downstream evaluation. Among those correlations, we found that Folding Barrier—the activation energy of refolding from the most stable RNA structure to the active gRNA structure—is the most important of those parameters for computational gRNA design. In our experience, Folding Barrier was useful enough that we could rely on it for the forward design of gRNA spacer sequences—and new CRISPRa promoters that match them—required for multi-gene CRISPRa expression control.

Once we understood gRNA folding well enough to design arbitrary numbers of new CRISPRa promoters, we built three gRNA-promoter pairs representing three orthogonal nodes of expression control. Again, the orthogonality comes from the sequence specificity of each gRNA’s spacer sequence. It is also known that tuning of CRISPR systems is possible by truncating the length of the spacer sequence [4], which in turn diminishes the energy of binding the CRISPR complex to the DNA. Therefore, a crucial advance was to implement this independent tunability in our three-promoter system, enabling control of enzyme stoichiometry within a pathway. All of the different combinations of enzyme expression levels represent a design space that often has a complex production landscape containing local maxima and minima. Experimentally exploring this space can reveal that production landscape and therefore reveal the most productive enzyme expression levels for each node, making an independently-tunable multi-gene expression system very useful for early-stage metabolic engineering.
This is when Ian jumped in to tackle an even more sophisticated metabolic engineering question: synthesizing human milk oligosaccharides (HMOs), a group of essential compounds found in breastmilk. Because human milk is sometimes in short supply, there is a need for alternative production of HMOs, e.g. LNT (lacto-N-tetraose), as infant formula additives. Indeed, we built this pathway in support of a project funded by a company (BASF) aiming to develop new microbial LNT production technology. The three-gene LNT pathway presented a great opportunity to apply our existing combinatorial CRISPRa technology. Especially because the synthetic part of the pathway intertwines with the host’s native carbohydrate metabolism, Ian thought that combinatorial gene expression profiling should provide us some insight into improving production. It turns out that LNT’s production landscape with our original three-gene pathway does not map smoothly to the design space available to our CRISPRa promoters. The combinatorial approach was able to identify a bottleneck within the pathway resulting from poor matching of enzyme activities. Therefore, replacing a single inefficient enzyme with a faster variant boosted the final LNT titer, as did fine-tuning the gene expression levels of three genes. Since we had a wide experimental profile of the LNT production landscape, we saw an opportunity to apply the data to train an ML model aimed at optimization of metabolic pathways.
Modeling approaches are an excellent opportunity to minimize the arduous and labor-intensive aspects of metabolic engineering, and we wanted to see how modeling could improve our approach beyond the experimental data we had gathered. Here, we collaborated with experts in machine learning for metabolic engineering, and used their Automated Recommendation Tool (ART) [5] to explore if we could gain additional understanding of expression profiles beyond the 64 variations that we had tested experimentally. This attempt provided two benefits: 1) increasing the resolution of our profiling of the expression space, and 2) decreasing the amount of experimental work required to understand the production landscape. We found that ART, when trained with our 64-sample LNT dataset, actually validated the aforementioned bottleneck, despite having no mechanistic understanding of the enzymes involved. Further, it made recommendations for new strains, clearly including the lessons learned from our experimental set. Finally, by training the model with variously-sized subsets of experimental data, we found an optimal tradeoff between experimental work and model accuracy, specific to this particular pathway. Extrapolating this concept beyond three-gene pathways, for instance to perhaps five, nine, even 25 genes, would help minimize experimental characterization of such a huge combinatorial design space, a very important task within the framework of Design-Build-Test-Learn (DBTL) cycles.
For a project that started from a desire to simply improve the predictability of gRNA folding, we find it remarkable how each stage of the project opened another door to new technologies in metabolic engineering: from gRNA design, through promoter design, through circuit design, to pathway design. Now that we have a variety of CRISPR tools developed and complemented by the modeling approaches, our ongoing effort in the lab is to apply the knowledge acquired to inform further development of our DBTL workflow, hopefully resulting in quicker completion of each DBTL cycle—and therefore a quicker route to better production strains. Please stay tuned for our upcoming papers!
References:
[1] Dong, C., Fontana, J., Patel, A., Carothers J. M., Zalatan, J. G. Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat Commun 9, 2489 (2018). https://doi.org/10.1038/s41467-018-04901-6
[2] Fontana, J., Dong, C., Kiattisewee, C., Chavali, V. P., Tickman, B. I., Carothers, J. M., Zalatan, J. G. Effective CRISPRa-mediated control of gene expression in bacteria must overcome strict target site requirements. Nat Commun 11, 1618 (2020). https://doi.org/10.1038/s41467-020-15454-y
[3] Alba Burbano, D., Cardiff, R. A. L, Tickman, B.I., Kiattisewee, C., Maranas, C.J., Zalatan, J. G., Carothers, J. M. Engineering activatable promoters for scalable and multi-input CRISPRa/i circuits. PNAS 120(30):e2220358120 (2023). https://www.pnas.org/doi/10.1073/pnas.2220358120
[4] Qi, L.S., Larson, M. H., Gilbert, L. A., Doudna, J.A., Weissman, J.S., Arkin, A. P., Lim, W. A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:(5):1173–83. (2013) https://doi.org/10.1016/j.cell.2013.02.022
[5] Radivojević, T., Costello, Z., Workman, K., Garcia Martin, H. A machine learning Automated Recommendation Tool for synthetic biology. Nat Commun 11, 4879 (2020). https://doi.org/10.1038/s41467-020-18008-4
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in