Predicting the Unbreakable: How AI and Density Functional Theory (DFT) are Designing PFAS-Destroying Catalysts
Published in Chemistry, Ecology & Evolution, and Materials
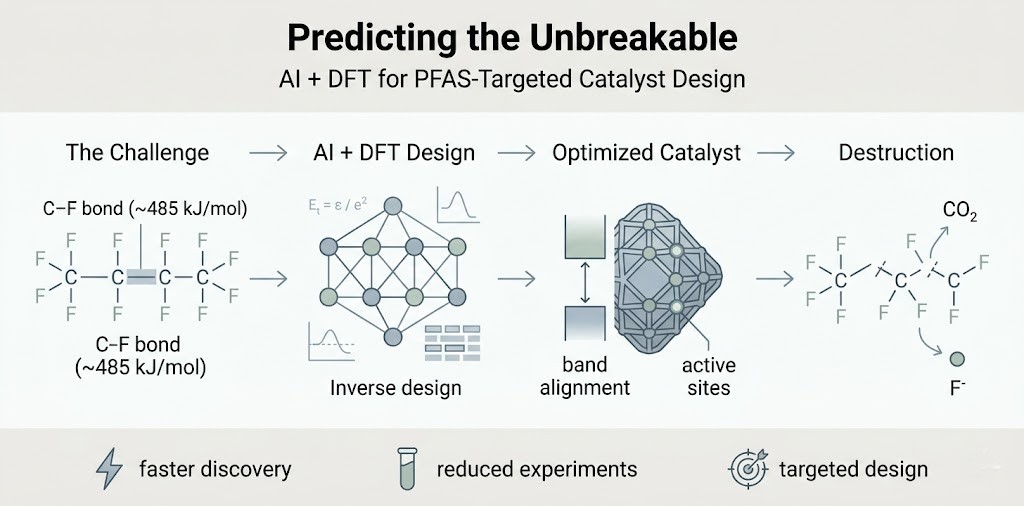
The Bottleneck of Empirical Chemistry
Historically, materials science has operated on a strict "synthesize, test, iterate" loop. While this empirical approach has yielded incredible environmental technologies, it is fundamentally bottlenecked by human limitations.
When attempting to design a catalyst to dismantle highly recalcitrant emerging contaminants—like per- and polyfluoroalkyl substances (PFAS)—the sheer number of variables is staggering. The possible combinations of transition metal dopants, oxygen vacancy densities, and heterojunction alignments in materials like spinel ferrites are nearly infinite. We can no longer afford the time, capital, or chemical waste required to synthesize and test thousands of random variations in a wet lab, hoping to stumble upon the perfect thermodynamic fit.
The Deep-Tech Pivot: Inverse Design
To secure our global water cycle, we must transition from reactive experimentation to predictive engineering. The frontier of this shift is Inverse Design.
Instead of building a material and observing its properties, inverse design flips the script. We start with the exact chemical behavior we require—for example, generating specific sulfate radicals or maintaining a highly negative conduction band edge—and let computational algorithms work backward to generate the atomic architecture needed to achieve it.
This is made possible by the powerful synergy of machine learning (ML) and Density Functional Theory (DFT). While ML algorithms can rapidly screen massive databases of known material properties, rigorous DFT calculations simulate the precise quantum mechanics and electron behaviors at the atomic level.

Targeting the C-F Bond In Silico
This computational approach is critical for "forever chemicals." The C-F bond in PFAS is one of the strongest in organic chemistry (~485 kJ/mol), making it incredibly resistant to conventional oxidative attacks.
By utilizing DFT, we can map the exact thermodynamic band alignments and charge-transfer pathways required to cleave this specific bond in a virtual environment. The algorithm can predict exactly which transition metal substitution (e.g., swapping nickel for zinc) or what specific degree of crystal lattice inversion will maximize the binding energy between the catalyst surface and the PFAS molecule, lowering the activation barrier before we ever synthesize the material.

The AI-to-Lab Pipeline: A Reality Check
As scientists, we must remain grounded: AI is an accelerator, not a magic wand. The computationally predicted structures must still be synthesized, characterized, and validated under real-world, complex wastewater conditions.
However, the efficiency gains are profound. Instead of synthesizing 100 blind variations, an environmental deep-tech lab can synthesize only the top three highest-probability candidates predicted by the algorithm.

Conclusion
The environmental engineers of the future will be just as proficient in Python and computational modeling as they are in analytical chemistry. By leveraging AI and DFT, we are no longer just searching for the perfect catalyst—we are coding it.
How quickly do you think computational materials science will become the standard baseline for all new environmental remediation grants? Let’s discuss in the comments below!
Dr. Akeem Adeyemi Oladipo is a globally recognized Research Professor of Materials and Environmental Chemistry at Eastern Mediterranean University. Ranked among the world's Top 2% Scientists, his expertise bridges nanotechnology, renewable energy, and electro-analytical chemistry. He specializes in advanced materials synthesis for wastewater treatment, highly sensitive electrochemical biosensors, and solar energy conversion. By integrating machine learning and artificial neural networks, his research drives scalable deep-tech innovations in environmental sustainability and energy storage.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in