The Molecular Imprinting 'Black Box': Why We Stopped Guessing and Started Calculating Sensor Selectivity
The Trial-and-Error Trap in Sensor Design
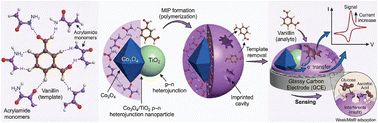
Molecular Imprinting Technology (MIT) is brilliant in theory. By polymerizing monomers around a target analyte and then washing the template away, we create recognition cavities with complementary shape and functional groups.
However, the reality of fabricating these sensors is often frustratingly empirical. For years, the interaction between the metal oxide support, the functional monomer, and the target analyte has been optimized through trial and error. This "black-box" design approach hampers rational sensor development. When a sensor failed in a complex food matrix, we rarely knew why at the atomic level.
With our latest work on vanillin detection—just published in Analytical Methods—my co-authors (Favour Ezinne Ogulewe, Mustafa Gazi) and I decided it was time to open the black box.
Building the Engine: The p-n Heterojunction
Before addressing selectivity, we needed raw electrocatalytic power. Pristine Co3O4 is a potent p-type semiconductor, but it suffers from aggregation and limited conductivity during prolonged operation.
By anchoring it onto n-type TiO2, we established a p-n heterojunction. The differing work functions drive Fermi level equilibration, creating a built-in internal electric field. This isn't just a structural support; it is a localized driving force that rapidly sweeps electrons away from the catalytic interface, drastically reducing charge-transfer resistance (by over 260-fold) and suppressing electron-hole recombination.
Replacing Guesswork with Density Functional Theory (DFT)
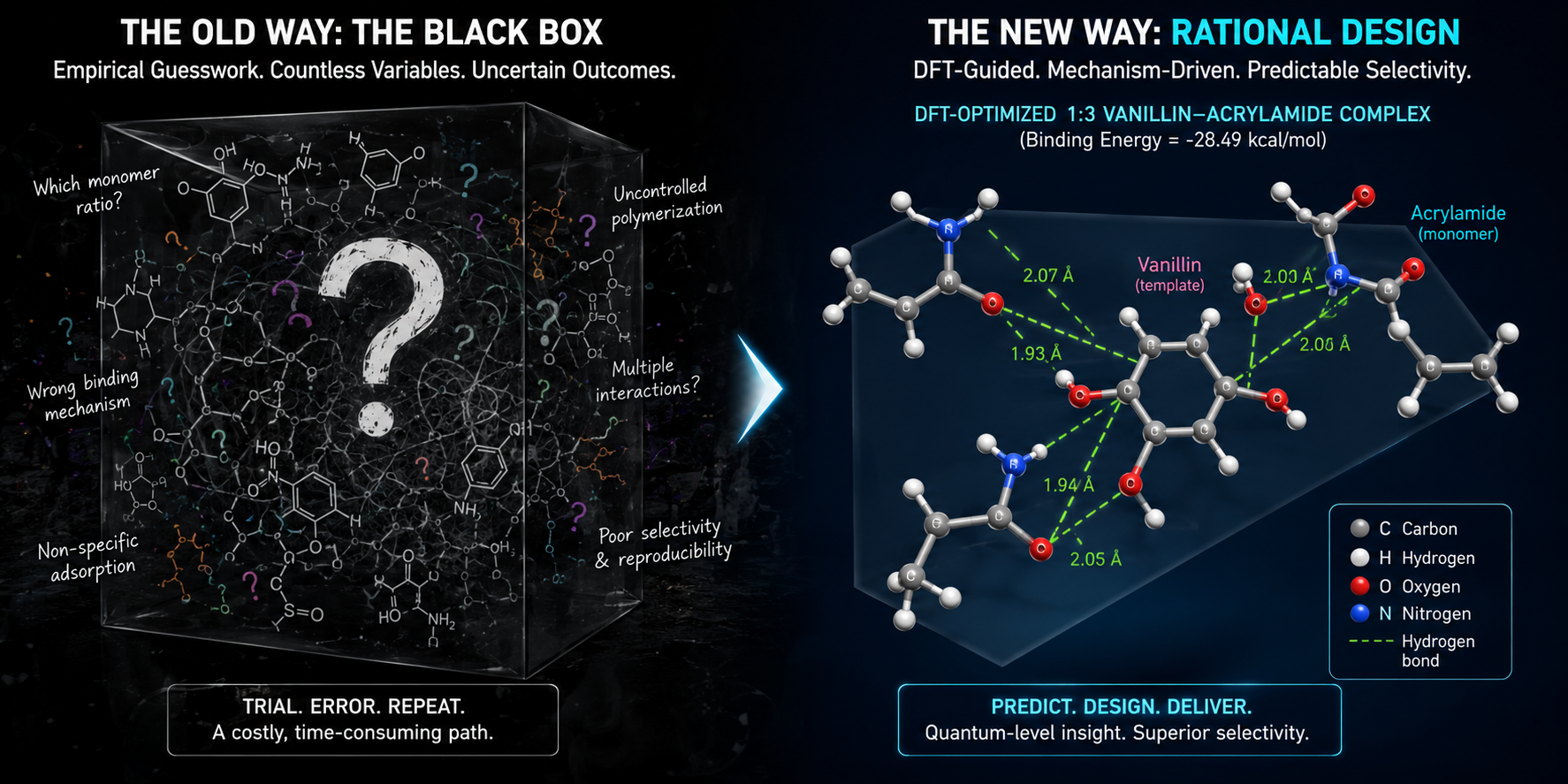
To build the recognition layer, we refused to guess the optimal monomer ratios. Instead, we used Density Functional Theory (DFT) and Monte Carlo simulations.
Our calculations revealed that binding strength doesn't just increase linearly with more monomer. The thermodynamic optimum is strictly a 1:3 vanillin-to-acrylamide ratio. At this exact stoichiometry, three acrylamide units cooperatively engage vanillin through multiple hydrogen bonds without introducing steric frustration, yielding a massive interaction energy of -28.49 kcal/mol.
Crucially, the computed binding energies for interferents like glucose (-12.57 kcal/mol) and ascorbic acid (-20.23 kcal/mol) were significantly weaker. We didn't just observe selectivity; we calculated the exact hydrogen-bond network that enforced it.
The Real-World Result
This direct correlation between quantum-level interactions and macroscopic electrochemical performance means DFT is a predictive tool, not just a descriptive add-on. By relying on calculated thermodynamics rather than empirical luck, we achieved a detection limit of 0.06 µM and successfully quantified endogenous vanillin in highly complex commercial matrices like milk, ice cream, and biscuits.
To the analytical chemistry community: Is it time journals start demanding computational validation for MIP designs, or does the cost of DFT modeling create an unfair barrier to publication? Let's discuss.
Dr. Akeem Adeyemi Oladipo is a globally recognized Research Professor of Materials and Environmental Chemistry at Eastern Mediterranean University. Ranked among the world's Top 2% Scientists, his expertise bridges nanotechnology, renewable energy, and electro-analytical chemistry. He specializes in advanced materials synthesis for wastewater treatment, highly sensitive electrochemical biosensors, and solar energy conversion. By integrating machine learning and artificial neural networks, his research drives scalable deep-tech innovations in environmental sustainability and energy storage.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in