The role of tyrosine hydroxylase-dopamine pathway in Parkinson's disease pathogenesis
Published in Chemistry
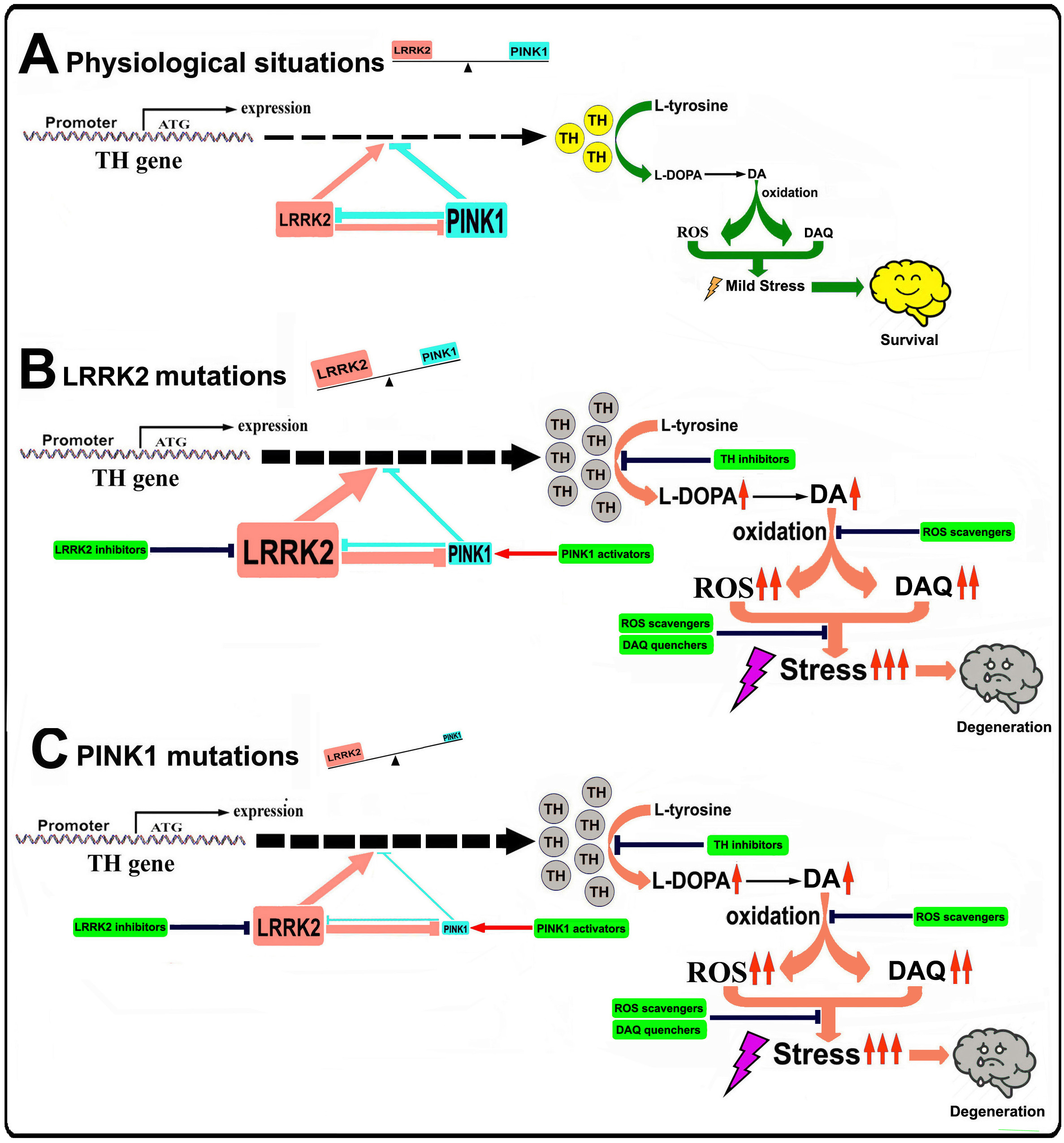

Most PD cases are sporadic, whereas monogenic forms of PD have been linked to multiple genes, including Leucine kinase repeat 2 (LRRK2) and PTEN induced kinase 1 (PINK1). The LRRK2 and PINK1 share some common functions as protein kinases that are involved in multiple signaling pathways. The LRRK2 is a large single polypeptide serine-threonine kinase protein containing multiple domains, while pathogenic LRRK2 mutations can increase LRRK2 kinase activity, contributing to DA neurotoxicity. The PINK1 is a 68 kDa serine-threonine kinase and its kinase activity is vital to its neuroprotective effects in dopaminergic neurons. Most PD associated PINK1 mutations are located in PINK1 kinase domain with impaired PINK1 kinase activity.
Our previous studies have demonstrated that endogenous DA and DA-dependent neurodegeneration have a pathophysiologic role in sporadic as well as familial form of PD. The DA toxicity is associated with iron species induced DA neuron demise. The DA neurotoxicity of wild type (WT) and mutant α-synuclein can be endogenous DA related. We showed that extra-mitochondrial WT PINK1 inhibit tyrosine hydroxylase (TH) and down regulate DA level in DA neurons, whereas pathogenic PINK1 mutations up-regulate TH activity and DA content in DA neurons. Furthermore mutant PINK1 induced DA neurodegeneration can be alleviated by a clinical grade drug, α-methyl-L-tyrosine (α-MT), a TH inhibitor.
We showed that LRRK2 could modulate TH activity and DA content in DA neurons in a LRRK2 kinase activity dependent manner. Increased LRRK2 activity can lead to up-regulated TH activity and enhanced DA content in DA neurons, while decreased LRRK2 activity can be associated with down regulated TH activity and DA level in DA neurons. In our transgenic (TG) Drosophila PD model PD-linked LRRK2 mutations disrupted the TH-DA pathway, resulting in up-regulation of DA level early in the disease which subsequently led to neurodegeneration. The LRRK2 induced DA toxicity and degeneration could be abrogated by WT PINK1 (but not PINK1 mutations) and early treatment with α-MT was able to reverse the pathologies in human neurons and TG Drosophila models with pathogenic LRRK2 mutations.
Our findings highlight the pathological roles of endogenous DA in PD pathogenesis as well as LRRK2-PINK1 kinase pair and TH-DA pathway as a potential targets in future anti-PD therapies. It will be interesting to investigate whether up-regulated TH-DA pathway functions and increased DA generation and oxidation can be identified in early stage of high-risk populations, such as LRRK2 or PINK1 mutation healthy carriers. Healthy subjects with pathogenic LRRK2 or PINK1 mutations identified with increased DA functions may be selected for neuroprotective drug trials.
In the current study, we showed that inhibition of TH by continuous low dose α-MT administration initiated at the early stage was able to prevent LRRK2 G2019S mutation induced DA neurodegeneration in our PD models. α-MT is an orally competitive TH inhibitor, which has been used clinically to treat hypertension-linked phaeochromocytoma and Dystonia, Dyskinesia and Huntington's disease. Low dose α-MT has been shown to be safe with no significant side effects even after prolonged use (3 years). Considering the excellent neurological pharmacology features with low toxicity and high human subject tolerance, the low dosage TH inhibitor therapy with α-MT seems to be a promising approach to protect DA neurons and prevent PD neurodegeneration. Early clinical trials in LRRK2 asymptomatic carriers can be a consideration.

Figure 1. Illustration of the role of LRRK2-PINK1 on TH expression and DA synthesis in DA neurons
Under physiological conditions, LRRK2 and PINK1 form a functional balance to maintain normal TH expression and DA synthesis in DA neurons. LRRK2 promotes TH expression and DA generation, while PINK1 suppresses TH expression and DA generation. LRRK2 and PINK1 can regulate degradation of each other, thus a balance can be reached. When LRRK2 is mutated its kinase activity is increased, leading to up-regulated TH expression and increased DA generation. Increased LRRK2 kinase activity can facilitate PINK1 degradation, down regulate PINK1 level and suppress PINK1 function. This will lead to imbalance between LRRK2 and PINK1, contributing to increased TH expression, enhanced DA generation, aggravated DA oxidation and elevated DA relevant stress in DA neurons, promoting neurodegeneration. When PINK1 is mutated, kinase activity will be impaired causing LRRK2-PINK1 imbalance and disrupting TH-DA pathway, promoting DA neuron vulnerability and neurodegeneration.
In our current study we demonstrated that in multiple in vivo and in vitro PD models, LRRK2 and PINK1 may form a functional protein kinase pair to modulate TH-DA pathway (Figure 1). The LRRK2 and PINK1 have opposing effects to modulate TH activity and DA content in DA neurons. Furthermore, LRRK2 and PINK1 facilitate proteasome degradation of PINK1 and LRRK2 proteins to reciprocally suppress their functions. A LRRK2-PINK1 physiologic balance on TH-DA pathway may be important for DA neuron viability (Figure 1). Under normal conditions, WT LRRK2 and PINK1 act together to maintain physiological TH and DA levels and promote DA neuron survival. However, when LRRK2 or PINK1 is mutated, the LRRK2-PINK1 balance will be disturbed, leading to DA neurodegeneration (Figure 1). Our findings provide support for potential clinical trials using TH-DA pathway inhibitors in early or prodromic PD.
I am a MD & Ph.D and a Clinician Scientist, working on translational neuroscience in National Neuroscience Institute of Singapore. My research expertise is on neurodegeneration in Parkinson's disease and other human neurodegenerative disorders, with specific focus on biomarkers and therapeutic targets for PD diagnosis and therapy.
Follow the Topic
-
Cellular and Molecular Life Sciences
Cellular and Molecular Life Sciences (CMLS) is a multidisciplinary Open Access journal covering the latest aspects of biological and biomedical research.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in