Uncovering a Non-Canonical Role of STING in Japanese Encephalitis Virus Infection
Published in Microbiology and Immunology
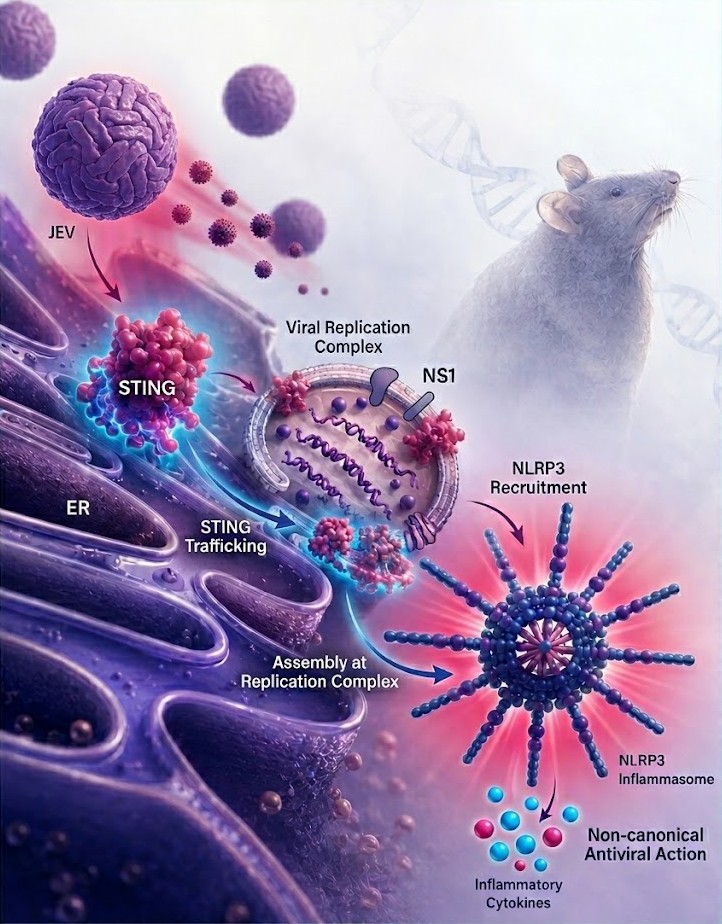
Every research project begins with a question. For us, that question was deceptively simple: how does STING contribute to host defense during Japanese encephalitis virus (JEV) infection?
JEV is a mosquito-borne neurotropic flavivirus and a leading cause of viral encephalitis in Asia, capable of causing severe neurological disease and long-term sequelae in survivors. Understanding the molecular mechanisms that govern host responses to JEV infection has been a long-standing focus of our laboratory, making STING an especially interesting molecule to investigate in this context.
STING is widely recognized as a central adaptor of the DNA-sensing pathway, orchestrating type I interferon responses against invading pathogens. However, JEV is an RNA virus. While several studies had reported STING activation during RNA virus infections, the biological significance and underlying mechanisms remained unclear. This apparent contradiction intrigued us and ultimately became the starting point of a journey that would span several years.
When we first began investigating STING in JEV infection, we expected to observe its classical antiviral functions through interferon signaling. Initial experiments indeed suggested that STING played a protective role during infection. However, as we delved deeper, the data began to tell a more complex story. Some of our observations simply could not be explained by the established paradigm of STING-mediated interferon responses.
At this point, the project entered what many researchers will recognize as the most challenging phase: when the data force you to abandon your original hypothesis.
One experiment led to another. Controls became new questions. Results that initially seemed contradictory began to fit together. We repeatedly tested whether the antiviral effect of STING depended on interferon signaling or autophagy, two pathways commonly associated with STING biology. To our surprise, the answer consistently came back no.
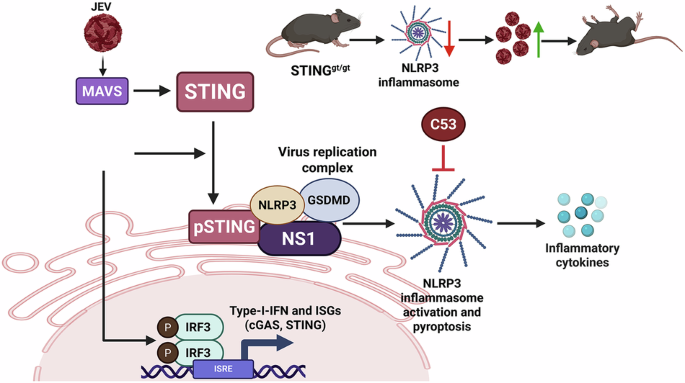
To gain an unbiased understanding of the host response during infection, we turned to RNA sequencing. The transcriptomic analyses revealed extensive changes in inflammatory pathways and provided important clues that could not be fully explained by canonical interferon signaling. What initially seemed like a secondary observation gradually evolved into the project's central theme. The emerging evidence suggested that STING was doing something unexpected—it was facilitating inflammasome assembly at the viral replication complex itself. This provided a completely different framework for understanding how STING restricts JEV replication.
Like most projects, this one was not a straight path from hypothesis to discovery. There were failed experiments, repeated optimizations, and long periods where progress felt frustratingly slow. Several key findings required extensive validation using different experimental approaches and model systems. In particular, studies using the Golden Ticket mouse model, which carries a loss-of-function mutation in STING, were critical in establishing the physiological relevance of our observations during JEV infection. Looking back, these challenges ultimately strengthened the study and increased our confidence in the conclusions.
This work benefited from the collective efforts of many researchers. The interdisciplinary nature of the project, spanning transcriptomics, molecular virology, innate immunity, and animal infection models, required integrating expertise across multiple approaches and experimental systems. These collaborative efforts helped transform a series of observations into a coherent mechanistic story. The project was funded by the Anusandhan National Research Foundation (ANRF), whose support was instrumental in enabling this work.
For us, this paper represents more than the discovery of a new antiviral mechanism. It highlights how much remains to be learned about innate immune signaling pathways, even for molecules as extensively studied as STING.
Science often advances not when experiments confirm what we expect, but when they challenge our assumptions. In our case, a DNA-sensing adaptor unexpectedly revealed a non-canonical antiviral function against an RNA virus. That surprise ultimately became the story.
We hope these findings stimulate further investigation into the diverse functions of STING and contribute to a better understanding of host–pathogen interactions during flaviviral infections.
I am a researcher with a strong interest in virology, host–pathogen interactions, and innate immune signaling. My work focuses on understanding how viruses manipulate cellular pathways to promote infection and how host immune mechanisms counter viral replication. I have experience with molecular and cellular biology techniques, animal models, viral pathogenesis studies, and immunological assays. My current research areas include flaviviruses, particularly Japanese encephalitis virus (JEV), the cGAS–STING pathway, antiviral immune responses, ISGs and virus-induced cellular remodeling. I enjoy discussing experimental design, data interpretation, scientific writing, and emerging developments in viral immunology and infectious disease research.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in