Why international collaboration is crucial for tracking antimicrobial resistance
Published in Microbiology

Antimicrobial resistance (AMR) is a global issue1. Previous studies have estimated 4.95 million deaths were associated with resistant infections in 20191, and this will be 10 million deaths per year by 20502. Extended-spectrum cephalosporins (ESC) are third- and four-generation cephalosporins categorised by the World Health Organization (WHO) as critically important antimicrobials for human and veterinary medicine3. Across the world, infections due to ESC resistant (ESC-R) bacteria are becoming difficult to treat due to their ability to produce extended-spectrum beta-lactamases (ESBLs) and plasmid-mediated AmpC β-lactamases (AmpCs)4, which are encoded by ESC-R genes.
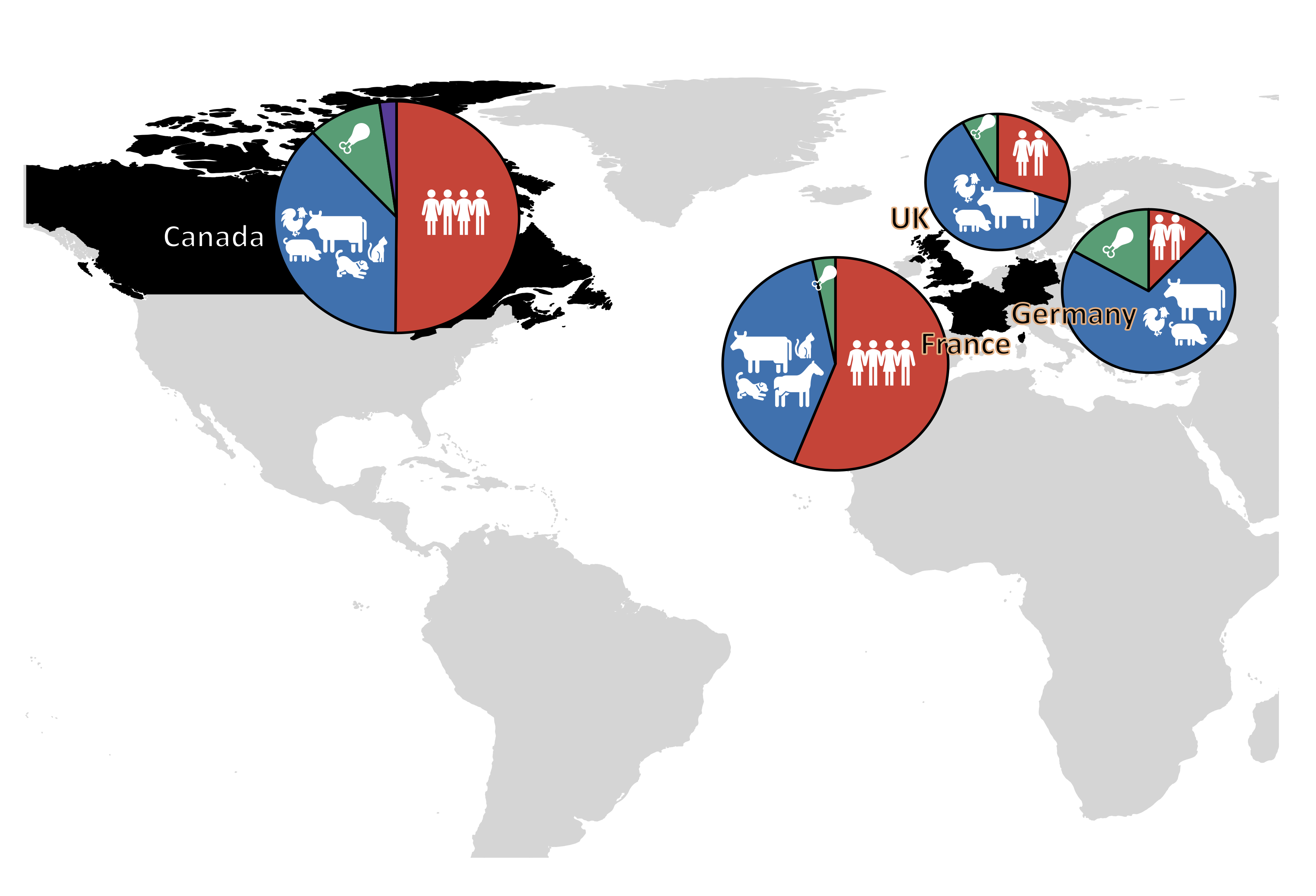
In this project we had the opportunity to work with key collaborators from multiple countries in Europe and North America to study AMR in bacteria from diverse sources of the various One Health compartments including humans, animals, food and the environment. This network of scientists from Canada, France, Germany and the UK brought collective expertise in genomics, microbiology, epidemiology and human and veterinary medicine. This network took time to build, building on existing relationships and developing new ones, where the partners’ commitment to collaborate and individual expertise were vital in the design and execution of this study.
The collaborative project was funded by the Joint Programming Initiative on Antimicrobial Resistance (JPIAMR; https://www.jpiamr.eu/). JPIAMR is an international collaborative platform constituted by 29 nations and the European Commission to combat AMR. The programme coordinates national research funding and supports international collaboration between researchers from different countries with diverse expertise to promote research on AMR in the One Health system and in a global context.
In this retrospective study we gathered whole genome sequences for a large collection of bacteria. Our strategy was to select isolates (bacteria) randomly from the large culture collections available through our collaborators across the three countries: Canada, France and Germany. This yielded 1,930 ESC-R bacterial genomes (1,524 from this study plus 406 from previous studies5–7) covering a time span of 15 years. Importantly, ESC-R isolates were selected on their phenotypic resistance to an ESC drug, and not on their genotype (i.e., presence of a specific ESC-R gene). Therefore, the relative frequency and occurrence of these genes were representative of what was circulating in the general populations during the study time frame and could be statistically compared. This strategy enabled comparative analysis of ESC-R genes between different countries and host species.
We faced some challenges with this approach, as some combinations of host species and country had relatively low numbers of genomes. We overcame this in two ways:
- We only made statistical comparisons of those compartments that had good representation, particularly for analysis of temporal trends in ESC-R.
- We used the strategy of taking multiple random subsets (without replacement) from compartments with larger sample sizes; these subsets were a similar size to smaller compartment, and the analysis was performed on these multiple subsets, ensuring comparison of like with like.
Another challenge that we faced was the reconstruction of plasmid sequences. With short-read data, plasmid sequences are generally fragmented into multiple pieces. We used multiple bioinformatics tools (MOB-suite8 and RFPlasmid9) to overcome this and predict the localization of ESC-R genes either in the plasmid or the chromosome. Moreover, we used long-read sequencing of a small subset to confirm plasmid prediction. Through analysis of a subset of typeable plasmids, we identified specific dominant plasmid types and subtypes associated with ESC-R genes in Escherichia coli and other bacterial species. Despite this, long-read sequencing at large scales is urgently needed for further in-depth investigation of AMR plasmids.
International collaboration allowed us to find that specific bacterial lineages, plasmid types and subtypes are the major drivers for the dissemination of ESC-R in E. coli worldwide. We learnt that plasmid subtypes could cross borders between different E. coli lineages, host species, countries and bacterial species. Our strategy can be applied to track pathogens as well as commensal bacteria such as E. coli to understand how AMR spreads.
International collaboration studies are essential to track bacterial AMR globally and to generate the essential knowledge needed to stop their spread.
Reference
- Murray, C. J. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
- O’Neill, J. Review on antimicrobial resistance. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. https://amr-review.org/sites/default/files/AMR Review Paper - Tackling a crisis for the health and wealth of nations_1.pdf (2014).
- World Health Organization. Critically important antimicrobials for human medicine. Ranking of medically important antimicrobials for risk management of antimicrobial resistance due to non-human use. 6th Revision 2018. (World Health Organization, 2019).
- Harris, P. Clinical management of infections caused by Enterobacteriaceae that express extended-spectrum β-lactamase and AmpC enzymes. Semin. Respir. Crit. Care Med. 36, 056–073 (2015).
- Pietsch, M. et al. Whole genome analyses of CMY-2-producing Escherichia coli isolates from humans, animals and food in Germany. BMC Genomics 19, 601 (2018).
- Kallonen, T. et al. Systematic longitudinal survey of invasive Escherichia coli in England demonstrates a stable population structure only transiently disturbed by the emergence of ST131. Genome Res. 27, 1437–1449 (2017).
- Ludden, C. et al. One Health genomic surveillance of Escherichia coli demonstrates distinct lineages and mobile genetic elements in isolates from humans versus livestock. MBio 10, 1–12 (2019).
- Robertson, J. & Nash, J. H. E. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genomics 4, 1–7 (2018).
- van der Graaf-van Bloois, L., Wagenaar, J. A. & Zomer, A. L. RFPlasmid: predicting plasmid sequences from short-read assembly data using machine learning. Microb. Genomics 7, (2021).
Acknowledgements: Special thanks to Professor Alison E. Mather (Head, Microbes in the Food Chain Institute Strategic Programme) and the Communications and Engagement team at the Quadram Institute Bioscience for the helpful suggestions whilst writing.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in