Widespread DNA off-targeting confounds RNA chromatin occupancy studies
Published in Biomedical Research
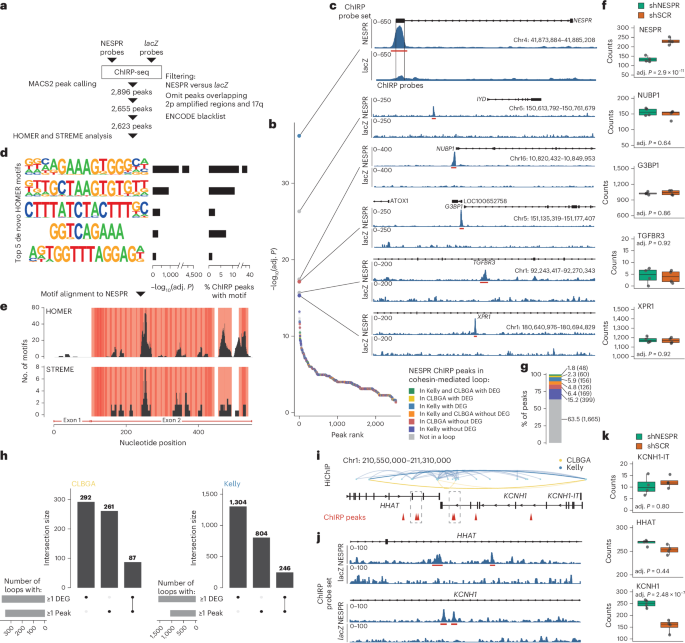
This project was conceived during my sabbatical at the Max Planck Institute for Molecular Genetics, hosted by Prof. Martin Vingron, a computational biologist. The Vingron lab conducted several studies that performed meta-analyses and comparisons of various tools for genomics research, which led me to think: What kinds of data or tools can be evaluated through such a lens in my field of long noncoding RNAs (lncRNAs)? The natural answer was datasets of lncRNA chromatin occupancy. Over the years, I’ve reviewed several manuscripts that included such experiments, and was often surprised by the discrepancy between the apparent low copy number of these RNAs in cells – often fewer than 5 copies per cell, and the reported number of genomic binding sites, often in the thousands, each of which seemed quite confident. When, as a reviewer, I raised questions about this apparent discrepancy, the reply was often citations of prior studies, using the same technique, which was most often ChIRP-seq, that also reported many binding sites for other non-abundant RNAs. Unfortunately, over the years, studies with such apparent discrepancies have accumulated. Importantly, the chromatin occupancy maps were often not the main experiment in these studies, but rather the last figure panel – the one aimed at connecting the phenotype observed upon lncRNA perturbation to the specific genes the lncRNA was suggested to regulate. The accumulation of such chromatin occupancy maps, dozens of which have been published in human and mouse cells, has fueled the notion that the task of ‘going to specific places in the genome’ is indeed something lncRNAs often carry out. Importantly, this was also a conceptually compelling model, as many chromatin-related complexes lack apparent sequence specificity yet regulate very specific genes across different developmental or physiological contexts. Recent studies have cast doubt on whether lncRNAs actually bind many of the complexes most often reported to act in this manner, such as Polycomb, but the evidence that lncRNAs often bind hundreds or thousands of discrete sites remains widespread in the literature.
During my sabbatical, I performed a rudimentary re-analysis of the data in some of the human studies, and after I returned to the Weizmann Institute, Micah Goldrich, a very talented PhD student in the lab, took over the project and injected substantial rigor and creativity into the analysis. Here, I need to explain shortly how the RNA chromatin occupancy are obtained. After cells are crosslinked, probes, typically short (~20 nucleotides), are used to pull on the RNA of interest, which is typically quite long (>1,000 nucleotides). DNA regions are extracted from the pulled-down material and prepared for sequencing. The recovered fragments are compared to an ‘input’ sample where the pulldown doesn’t take place. Together with Micah, we first found that the reported binding sites very often shared short sequence matches with the probes used in the pulldown experiments. For short matches of 7-12 nt or so, the number of sites that had such overlaps greatly exceeded (often by >10-fold) that expected by chance. While such matches might have been expected if the RNA of interest formed Watson-Crick base pairs with the target DNA, we found they occurred only in regions of the RNA bound by the probes, not elsewhere. This formed strong evidence that the probes, which were supposed to enrich the RNA, were instead directly enriching DNA fragments in an RNA-independent manner. Micah then cracked the mechanism through which this occurred – he found strong evidence that the probe matches were preferentially found at the ends of the sequenced reads, suggesting that the probes bound to single-stranded DNA fragments that can be formed during the process of library preparation.
After I presented our initial findings at the Noncoding Genome meeting at EMBL in October 2023 (a virtual presentation as I could not travel because of a war that had just broken out in Israel), I was contacted by Louis Delhaye and Pieter Mestdah from Ghent, who independently reached a similar conclusion. They studied a specific lncRNA, NESPR, and found that while it regulated the expression of a large number of genes, these genes were unrelated to the places where the lncRNA seemed to bind in the genome, and more concerningly, the bind sites reported by ChIRP-seq peristed even in cells where the lncRNA was not expressed at all, or in cells that did express the lncRNA but that were treated with RNase that was supposed to eliminate any RNA-related signal. We then decided to team up and present our independent findings in a joint manuscript that was a real pleasure to prepare and publish.
Beyond the findings on specific lncRNAs that our results cast a shadow on, our findings have broader implications for understanding how lncRNAs function, as they widen the gap in understanding whether lncRNAs, beyond a few well-studied examples such as Xist and Rox, indeed often target large numbers of distal chromatin sites. The mechanisms proposed for such targeting, such as triplex formation, have difficulty explaining how targeting specificity arises. We propose both computational and experimental methods to increase rigor in studying RNA-chromatin interactions, aiming to yield cleaner future maps that can expedite the study of lncRNA modes of action.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in