Behind the Paper: Catalyst self-assembly accelerates bimetallic light-driven water splitting
Published in Chemistry
Synthetic fuels would be attractive alternatives to fossil fuels, if chemists could devise methods that convert abundant resources into valuable fuels using renewable energy inputs. One promising strategy for synthetic fuel production is to use sunlight to generate hydrogen gas from water. Now, molecular catalysts have been found to self-assemble into nanoscale aggregates using noncovalent interactions that enable efficient photoelectrochemical H2 evolution from water (1).
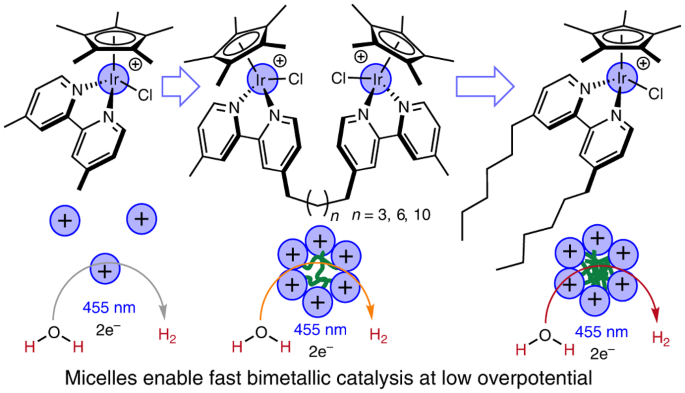
The molecular catalysts are rare examples of systems in which a single transition metal complex functions both as light harvesters and as fuel-forming catalysts (2). The original catalysts were organometallic iridium complexes with pentamethylcyclopentadienyl and bipyridine supporting ligands, similar to Ir-Me2 in Figure 1 (3). In prior work, the mechanism of catalysis was proposed to a involve bimetallic H–H coupling step (4), which led us to design catalysts in which two iridium centers are covalently tethered to one another in the present study. Although initial studies showed faster rates, subsequent mechanistic studies demonstrated that the improved performance was not due to intramolecular H–H coupling as originally expected. Instead, the hydrophobic linkers serendipitously led to aggregation into micelles built entirely of catalysts. These catalyst-rich particles suspended in the aqueous medium had a rare combination of features: milder reduction potentials due to the many proximal cationic centers making each complex easier to reduce, and faster rates of catalysis ascribed to proximal catalysts ready to form H–H bonds between near neighbors.
Figure 1 shows the various catalyst structures that were compared and illustrates the principle that larger aggregates with more tightly packed iridium complexes will lead to faster catalysis and at milder applied potentials. This relationship is reflected in the graph comparing catalytic activity (kmax, the maximum rate constant for catalysis that was observed under the specific illumination and reaction conditions) and the overpotential (applied potential beyond the thermodynamic potential for water reduction to H2 that was needed to reach a particular activity).

Figure 1 Caption. Iridium catalysts studied in this work and illustration depicting the change in self-assembly as correlated with catalytic activity (top) and activity vs overpotential plot at 1 mM normalized Ir concentration showing no linear relationship and aggregating examples with higher rates and lower overpotentials than Ir-Me2. Activity is reported as kmax, the maximum achieved rate constant under the conditions of 455 nm LED illumination, pH 7 sodium phosphate buffer using chronoamperometry at the given overpotential.
The research was performed at the University of North Carolina at Chapel Hill. The work was started by an undergraduate student researcher all the way back in 2016, based on the hypothesis that linking catalysts would enable fast intramolecular coupling. The first systems developed had very long linkers, with eight or twelve carbons, thinking that flexibility would be key. A postdoctoral scholar arrived and noticed initial evidence of aggregation, leading to a thorough study using multiple spectroscopy and microscopy methods. We realized that the aggregation might follow the same principles discovered by Professor Xi-Kui Jiang of Shanghai Institute of Organic Chemistry in the 1980s regarding aggregation and self-coiling of organic molecules in aqueous solution (5). This helped inspire the design of catalysts like those with shorter five-carbon linkers and monometallic catalysts with varying substituent structures (featuring combinations of methyl, hexyl, and docecyl groups). As predicted, the catalysts that aggregate well give excellent catalytic performance, validating the hypotheses of the work. The COVID-19 pandemic temporarily interrupted the work, but after the labs reopened a master’s student and new postdoctoral scholar led efforts to quantify all catalytic activity and further develop the details of the mechanistic understanding.
The primary outcome of the work is insight into design principles for bimetallic catalysts. We find that judicious incorporation of hydrophobic groups can lead to self-assembly of catalysts into micelles that hold many active sites in close proximity. Any catalyst that features a bimetallic step might be expected to benefit from this design. The ability to work in water as a solvent may also be generally desirable, and it is a required feature of this strategy.
Acknowledgements: This work was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under award DE-SC0014255.
References
(1) Cloward, I.N., Liu, T., Rose, J. et al.Catalyst self-assembly accelerates bimetallic light-driven electrocatalytic H2 evolution in water. Chem. (2024). https://doi.org/10.1038/s41557-024-01483-3
(2) Brereton, K.; Bonn, A. G.; Miller, A. J. M. Molecular Photoelectrocatalysts for Light-Driven Hydrogen Production. ACS Energy Lett. 3, 1128–1136 (2018). https://doi.org/10.1021/acsenergylett.8b00255
(3) Pitman, C. L.; Miller, A. J. M. Molecular Photoelectrocatalysts for the Visible Light-Driven Evolution of Hydrogen from Neutral Water. ACS Catalysis4, 2727–2723 (2014). https://doi.org/10.1021/cs500441f
(4) Chambers, M. B.; Kurtz, D. A.; Pitman, C. L. et al. Efficient Photochemical Dihydrogen Generation Initiated by a Bimetallic Self-Quenching Mechanism. Am. Chem. Soc. 138, 13509–12512 (2016).https://doi.org/10.1021/jacs.6b08701
(5) Jiang, X.-K. Hydrophobic-Lipophilic Interactions. Aggregation and Self-Coiling of Organic Molecules Chem. Res. 21, 362–367 (1988). https://doi.org/10.1021/ar00154a002
Follow the Topic
-
Nature Chemistry

A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.
Your space to connect: The Fuel cell technologies Hub
A new Communities’ space to connect, collaborate, and explore research on Electrochemistry, Chemical Engineering, and Fuel Cells!
Continue reading announcement

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in