CAR-MSCs: the New Drivers of Immune System Regulation
Published in Bioengineering & Biotechnology and Biomedical Research
Autoimmune and immune-mediated diseases are affecting a growing proportion of the population worldwide, and the complex, chronic nature of these diseases leads to significant health and financial burden in patients, caregivers, and healthcare systems. Therapeutic options have been refined through a better mechanistic understanding of the immune system; however, treatment options for refractory disease remain very limited. Many current treatment options are given continuously, and attempt (and often fail) to control symptoms rather than address the root cause of the issue.
Mesenchymal stromal cells (MSCs) are multipotent, “stem-like”, progenitor cells that mediate immune cell homeostasis in the body and can be isolated from stromal compartments including adipose tissue and bone marrow. These readily available cells are strong candidates for cellular therapeutics due to their significant regenerative and immunomodulatory properties and their reduced immunogenicity allowing for potential as an off-the-shelf cellular therapy1. MSCs have been widely investigated as a therapeutic option for immune-mediated diseases including Graft vs. Host Disease (GvHD). Despite the safety of MSC therapy being tested and verified across thousands of past and ongoing clinical trials, MSC therapeutic efficacy has been disappointing. Existing MSC therapeutic approaches are suboptimal, and mechanisms for the lack of efficacy are in large part due to insufficient immunosuppression exerted by the MSC and limited trafficking to relevant tissues following MSC therapeutic administration1, 2.
The application of synthetic biology and cellular engineering to MSCs is an attractive approach to overcome the current limitations of MSC therapies. Such improvement of cellular therapies has been achieved with resounding success in the case of chimeric antigen receptor T (CART) cells in hematological malignancies3. A CAR is a synthetic receptor protein that redirects cells to targets via an antigen-binding single chain variable fragment (scFv) and enhances cell functional activation via intracellular signaling domains4. The ensuing clinical success of CART cells has caused a paradigm shift in the use of synthetic biology as it applies to cell therapy augmentation5.
The success of CART cell therapy in the treatment of cancer provides a strong rationale to apply CAR technology to the field of MSC therapies. Here, we proposed we could enhance MSC trafficking and immunosuppressive properties and hence broaden MSC applications in immune diseases through design and incorporation of CARs. We sought to improve MSC therapy for immune-mediated diseases by engineering MSCs with CARs. We hypothesized that, compared to unmodified MSCs, we could design CAR-MSCs with enhanced trafficking to target sites through the CAR scFv design and strengthen immunosuppressive activation through incorporation of specific intracellular signaling domains.
To test our hypothesis, we generated a proof-of concept CAR-MSC design which incorporated an scFv that would bind to target protein, E-cadherin, which is highly expressed on colon tissue. E-cadherin is often a target for overactive T cells in immune disease, and therefore lead to extensive colon damage for affected patients. We also incorporated a functional CD28ζ intracellular signaling domain into the CAR in hopes that upon interaction with damaged E-cadherin+ tissue, our CAR-MSCs would be stimulated to exert enhanced immunosuppressive function (Fig. 1).

Figure 1. Design of CAR MSC. Mesenchymal stromal cells expressing CAR receptor to enhance their trafficking through the scFv and their suppressive functions through the intracellular signaling domains. Created with BioRender.com
Following design of our CAR, we had to ensure that CAR could be stably delivered or transduced to MSCs without altering their stem cell-like properties. While primary T cells are relatively easy to transduce, it took some trial and error to get strong CAR transduction on MSCs. Eventually, we discovered that the addition of protamine sulfate yielded high, stable CAR expression on MSCs and created the first CAR-MSCs (Fig.2). We also confirmed that CAR-MSCs maintain their undifferentiated stem-like phenotypes through surface marker expressing and genetic studies, easing concerns that genetically modifying the MSCs could result in unwanted changes to their properties.
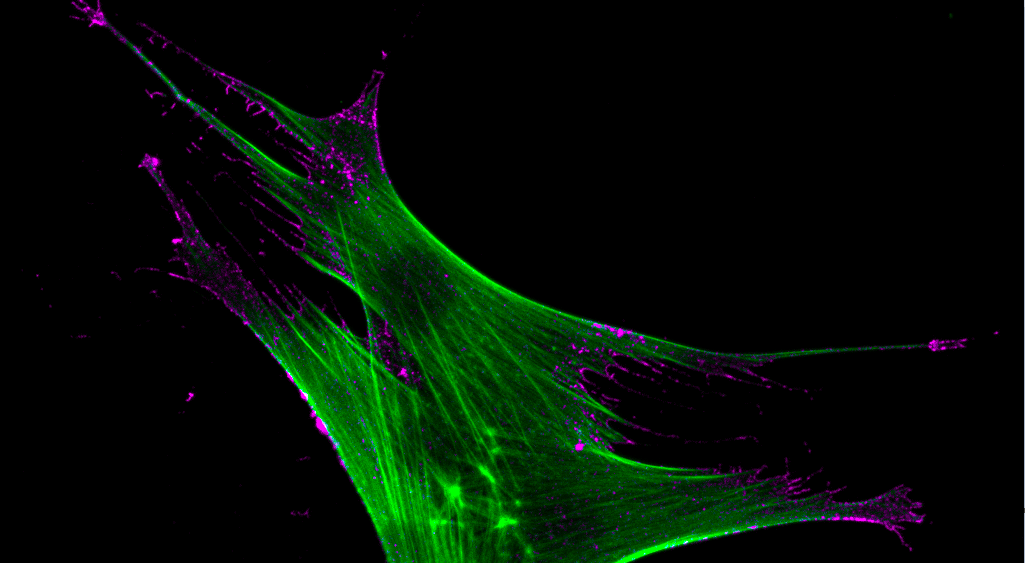
.png "Figure 2. CAR MSC. Mesenchymal stromal cells expressing CAR receptor at the cell surface, where it detects and engages with target cells to initiate immunosuppressive response. The CAR MSC actin-cytoskeleton is shown in green, and chimeric antigen receptor is shown in purple.")
Figure 2. CAR MSC. Mesenchymal stromal cells expressing CAR receptor at the cell surface, where it detects and engages with target cells to initiate immunosuppressive response. The CAR MSC actin-cytoskeleton is shown in green, and chimeric antigen receptor is shown in purple.
We next set out to determine if CAR-MSCs showed enhanced immunosuppression compared to unmodified MSCs. We tested CAR-MSCs in a preclinical mouse model of graft-vs-host disease (GvHD) and found significant therapeutic benefits over unmodified MSCs, including lower GvHD clinical symptom score and prolonged survival.
Given these striking results, we dove into potential mechanisms for CAR-MSC functions. In our mouse models, we saw reduced numbers of circulating human T cells, increased percentage of immunosuppressive regulatory T cells, and increased serum levels of immunosuppressive cytokines after CAR-MSC infusion compared to control MSCs . Our results revealed enhanced CAR-MSC trafficking to relevant target tissues (in this case, E-cadherin+ colonic tissue). Enhanced accumulation in target tissues was not seen with CAR-MSCs targeting an irrelevant antigen, indicating that the specificity of the scFv to a disease-specific antigen was crucial to our observations of improved CAR-MSC trafficking.
Having demonstrated the importance of the extracellular antigen-binding domain of the CAR, we also assessed the impact of the intracellular CAR signaling domains. In our GvHD mouse models, we discovered that CAR-MSCs lacking one or more intracellular signaling domains resulted in inferior therapeutic effects. Through RNA sequencing, we found that antigen-stimulated CAR-MSCs upregulate several pathways relevant to immunosuppression compared to unmodified MSCs or unstimulated CAR-MSCs indicating functional activation.
In this study, we have demonstrated the feasibility of producing CAR-MSCs, discovered some of the mechanisms behind their enhanced immunosuppressive capabilities, and have shown that this approach is safe and effective in suppressing aberrant T cells with enhanced therapeutic efficacy in animal models6. Given the modular nature of CARs, CAR-MSC therapy can be tailored for a variety of disease settings. We are optimistic that CAR-MSCs will show improved therapeutic benefits in patients with various immune-mediated diseases and can be governed by alternative disease-specific. We hope this work serves as a proof of concept for future CAR-MSC applications to improve the treatment of various immune-mediated and degenerative diseases.
References
- Galipeau, J. & Sensebe, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 22, 824-833 (2018).
- Levy, O. et al. Shattering barriers toward clinically meaningful MSC therapies. Sci Adv 6, eaba6884 (2020).
- Kenderian, S.S., Ruella, M., Gill, S. & Kalos, M. Chimeric antigen receptor T-cell therapy to target hematologic malignancies. Cancer Res 74, 6383-6389 (2014).
- Sakemura, R. et al. Development of a Clinically Relevant Reporter for Chimeric Antigen Receptor T-cell Expansion, Trafficking, and Toxicity. Cancer Immunol Res 9, 1035-1046 (2021).
- Sakemura, R.L. et al. CD19 occupancy with tafasitamab increases therapeutic index of CART19 cell therapy and diminishes severity of CRS. Blood 143, 258-271 (2024).
- Sirpilla, O. et al. Mesenchymal stromal cells with chimaeric antigen receptors for enhanced immunosuppression. Nat Biomed Eng (2024).
Follow the Topic
-
Nature Biomedical Engineering

This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.
Related Collections
With Collections, you can get published faster and increase your visibility.
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in