Cobalt(III) N-enolate carbene radicals with a protective function
Published in Chemistry

Researchers of the Van 't Hoff Institute for Molecular Sciences at the University of Amsterdam (UvA) have revealed that porphyrin ligand modification by acceptor-acceptor iodonium ylides ‒ reactive carbene transfer agents that can easily lead to “over-carbenation” of a catalyst ‒ is not necessarily a dead-end catalyst deactivation pathway, but can actually have a useful protective function by slowing-down deactivation pathways of the metalloradical cobalt(II)-tetraphenylporphyrin catalyst. This enables effective and high yielding cyclopropanation reactions with acceptor-acceptor iodonium ylides at room temperature.
Epping et al. investigated cobalt(II)-catalyzed cyclopropanation reactions using iodonium ylides as disubstituted acceptor-acceptor carbene precursors. Cyclopropanes are important substructures in specific medicines and several other bioactive molecules, but synthesis of disubstituted acceptor-acceptor cyclopropanes under mild conditions using base-metal catalysts is still a largely unsolved problem, thus limiting synthetic applications. This is in part due to the fact that diazo precursors that do allow generation of carbenes with two acceptor substituents are typically difficult to activate by commonly applied base-metal catalysts. The researchers in Amsterdam therefore took a different approach, and investigated the use of iodonium ylides instead, which are more powerful carbene delivering precursors than diazo compounds. However, precisely because of their enhanced reactivity, iodonium ylides are also well-known to trigger “over-carbenation” of the catalyst. With too many carbene units generated at the catalyst ligand modification and active site blocking is often observed, with catalyst deactivation typically being the end-result. Indeed, for several closed-shell porphyrin complexes, Mansuy and coworkers have reported already in the 1980’s that formation of N-enolates bridging between the metal and a pyrrole unit of the porphyrin ligand not only results in transition metal complexes with a strongly modified porphyrin ligand, but which are also typically completely inactive in carbene transfer catalysis. However, the present study shows that this is not general and the result apparently depends strongly on the electronic structure of the catalyst. The authors have shown that for paramagnetic metalloradical cobalt(II) porphyrin complexes N-enolate formation does not lead to catalyst deactivation at all, but is in fact beneficial. Due to the unique electronic structure of these catalysts, N-enolates are in equilibrium with carbene radical intermediates, and hence their formation is reversible. The N-enolate even has a protective function, slowing down catalyst deactivation via hydrogen atom transfer (HAT) from the solvent. The study thus reveals a unique interplay between the reactivity of hypervalent iodonium ylides and the open-shell metalloradical cobalt(II)-tetraphenylporphyrin complex in catalytic cyclopropanation reactions, enabling fast and high-yielding reactions at room temperature.
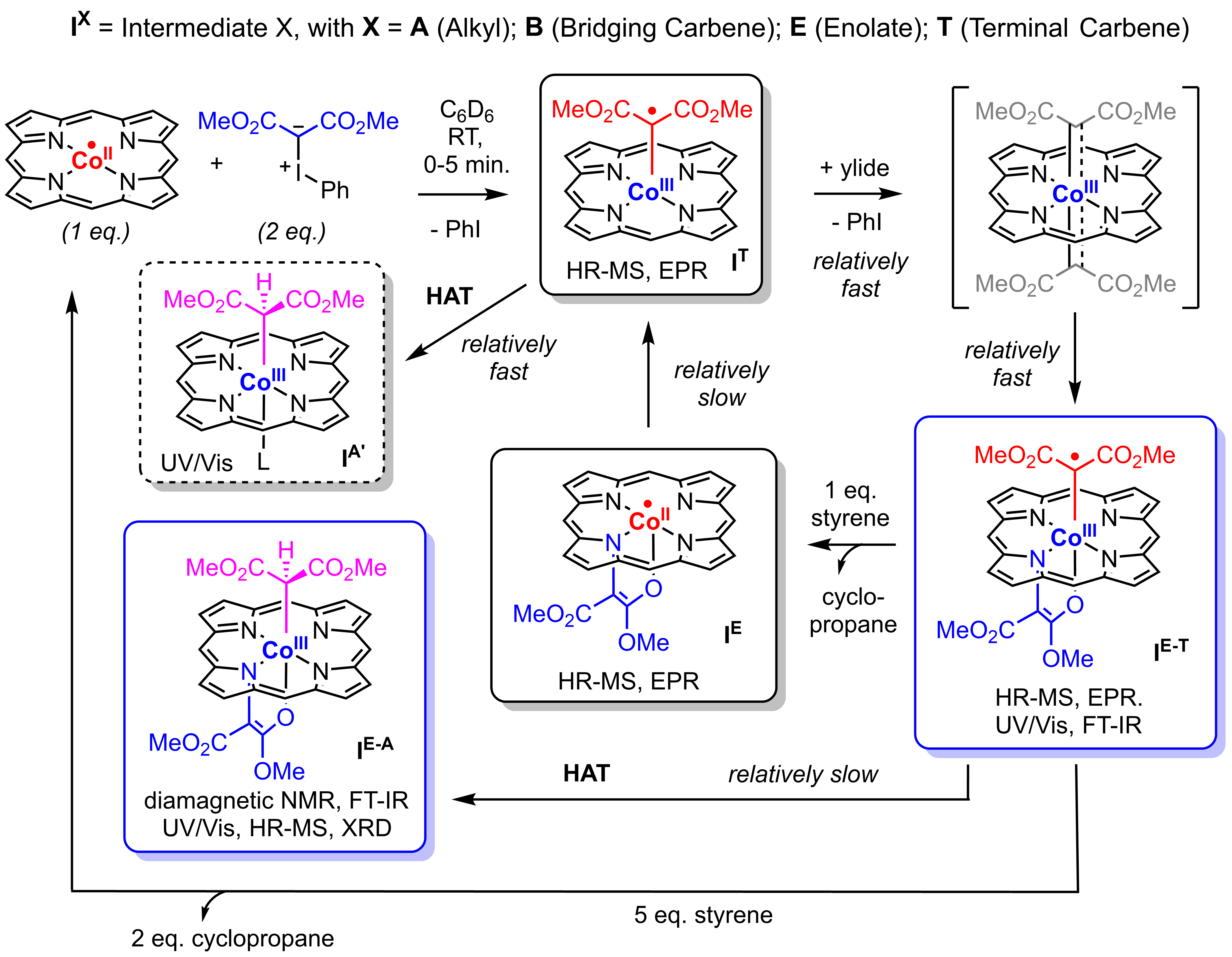
Both “mono-carbenoid” (cobalt(III) mono-carbene radical IT) and “bis-carbenoid” (cobalt(III) N-enolate carbene radical IE-T) species are detected under the catalytic reaction conditions. Epping et al. have shown that catalytic transfer of both “carbenoid” moieties of IE-T to C=C bonds is possible, which proceeds via a complex catalytic mechanism involving two interconnected cycles. The reactive paramagnetic cobalt(III) mono-carbene radical (IT) and cobalt(III) N‑enolate, carbene radical (IE-T) intermediates involved in these reactions are characterized using a combination of several spectroscopic/spectrometric techniques, experimental design, computational modelling and trapping experiments, including X‑ray diffraction of a HAT deactivated N-enolate complex. Remarkably, the “mono-carbenoid” species IT do not form stable N‑enolate isomers, while the “bis-carbenoid” species are most stable as cobalt(III) N-enolate carbene radical species IE-T, and as a result N‑enolate formation is reversible. Since the “bis-carbenoid” cobalt(III) N-enolate carbene radical complex IE-T has a reduced tendency to undergo catalyst deactivation via HAT when compared to the “mono-carbenoid” species IT, the observed “over-carbenation” by iodonium ylides is actually beneficial for catalysis. The interplay of opposing stabilities between terminal and N‑bridging carbene radical moieties dictated by the CoII/CoIII-redox cycle thus plays an important role, and explains the unique active participation of the otherwise catalytically inactive N-enolate species. Future application of these unique species holds the potential to unlock new scaffolds via the superior activation of acceptor-acceptor iodonium ylides over diazo compounds.
Follow the Topic
-
Nature Chemistry

A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in