DNA methylation heterogeneity and epipolymorphism in clear cell kidney cancer
Published in Cancer, Genetics & Genomics, and Biomedical Research
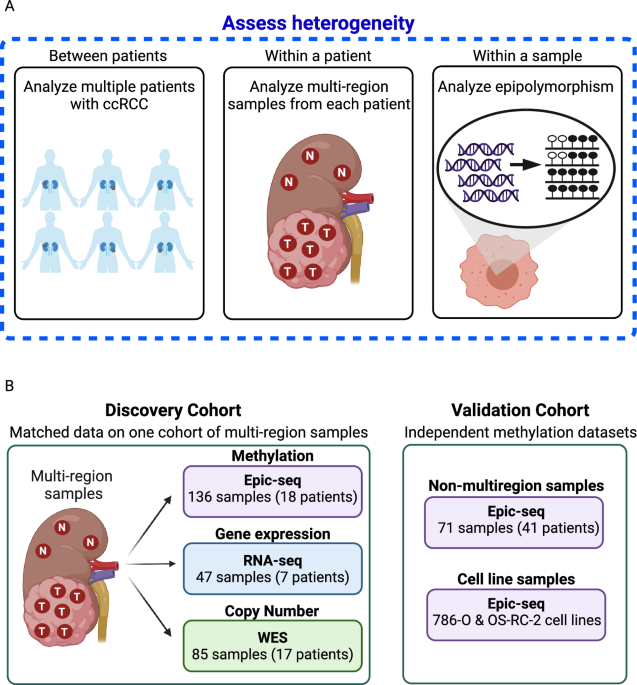
In our study we focused on the DNA methylation in clear cell renal cell carcinoma (ccRCC). We investigate genetic and epigenetic heterogeneity and evolution of ccRCC by combining whole-exome sequencing of multiple samples per patient with bisulfite sequencing of the same samples (Figure 1).
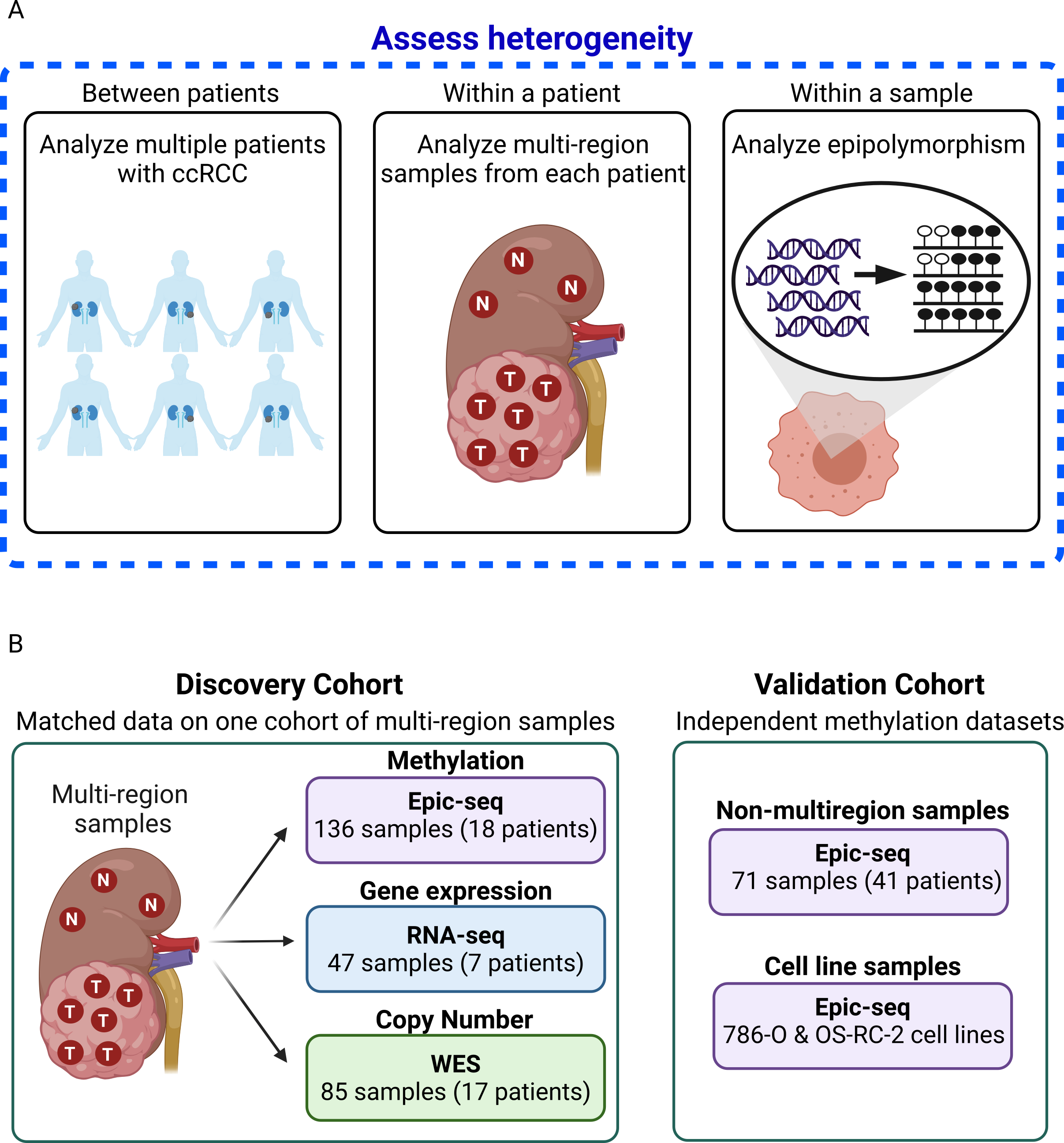
Figure 1: Schematic representation of samples and analysis
Studying DNA methylation with regard to tumor evolution and heterogeneity comes with several advantages: First, methylation is a conserved and stable DNA modification in vertebrates typically at CpG dinucleotides that can be easily detected by probe (array)- or sequencing based methods. Second, DNA methylation usually represents an early event in tumorigenesis that is propagated to daughter cells[1]. Third, non tumor associated DNA methylation has a cell type specific pattern therefore allowing for cell type identification and data deconvolution.
Challenges in analyzing DNA methylation are mainly based on the large amount of around 30 million CpG dinucleotides in the human genome and the limited understanding of DNA methylation function outside of CpG rich islands in gene promoters. In addition, the methylation status of neighboring CpGs is not independent and deviates from equilibrium frequency which is important to account during data analysis [2]. For our study we have chosen a sequencing technique called EPIC-seq that targets a subset of around 3.3 million CpGs selected based on known functional relevance and emerging interest identified in previous methylation studies.
In contrast to an array based approach our targeted sequence level based methylation analysis allowed not only to assess methylation differences at CpGs in different samples but also epipolymorphism in consecutive CpGs on different sequencing reads.
Epipolymorphism is defined as the probability that two epialleles randomly sampled from a DNA methylation locus (meaning a defined number of consecutive CpGs on the DNA sequence) differ from each other, although the mean methylation level of that locus is constant (Figure 2A)[3].
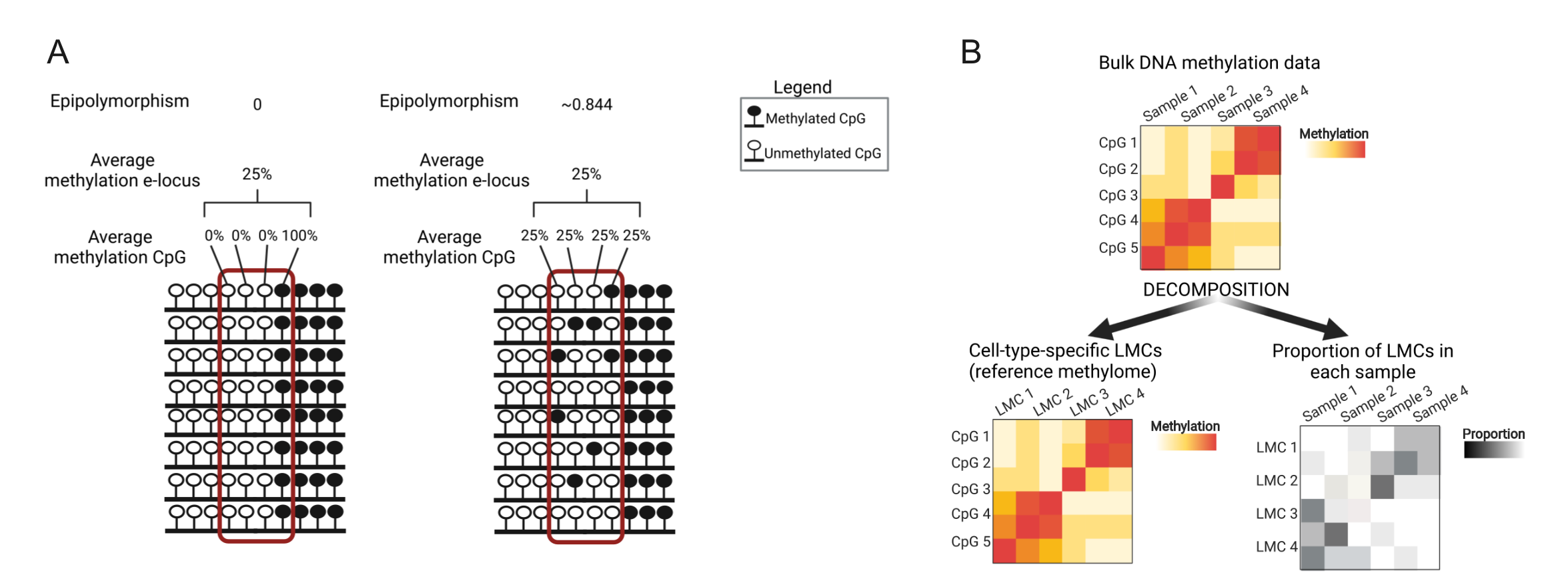
Figure 2: A) Schematic explanation of epipolymorphism and average methylation, adapted from [3]. B) Schematic explaining how bulk DNA methylation data are decomposed into cell-type-specific latent methylation components (LMCs) and the proportion of LMCs in each sample.
This led to our main finding of differential epipolymorphism at loci of 4 adjacent CpGs between ccRCC and normal tissue, in the promoter region of genes which are known to be associated with ccRCC. We also show an impact of differential epipolymorphism in gene promoters being an independent predictor of associated gene expression.
Our study design of multiregional ccRCC sampling and multi-omics sequencing allowed us to investigate tumor phylogenies either based on somatic copy-number alteration and DNA methylation data in 8 different patients. While the resulting phylogenetic trees of two patients suggested a co-evolution of DNA methylation and somatic copy-number alterations, the results for three patients were more diverse and for another three patients distinctly different, pointing to a separate evolution in these cases.
Using the cell-type specific patterns of DNA methylation to deconvolute the data we could identify a latent methylation component likely representing tumor infiltrating T-cells (Figure 2B). Levels of this component were higher in tumors with positive predictive parameters (lower grade, stage, Leibovich score, absence of necrosis, and did not develop a recurrence).
Overall, our study was able to shed light on the heterogeneity of DNA methylation in ccRCC in different facets. Future research should investigate epipolymorphism across different stages of ccRCC to determine whether evolutionary changes are associated with disease progression. Moreover, further research is needed to characterize and confirm the association between high T-cell infiltration in ccRCC and patient prognosis.
[1] Baylin, SB & Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends in Genetics. 2000. 13(4):168-174
[2] Saito, D. & Suyama, M. (2015). Linkage disequilibrium analysis of allelic heterogeneity in DNA methylation. Epigenetics, 10(12): 1093–1098.
[3] Landan G, Cohen NM, Mukamel Z, Bar A, Molchadsky A, Brosh R, et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet. 2012;44:1207–14
Follow the Topic
-
Oncogene
This journal aims to make substantial advances in our knowledge of processes that contribute to cancer by publishing outstanding research.






Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in