Exploring the role of nonlocal Coulomb interactions in perovskite transition metal oxides
Published in Materials
Theoretical Challenges in Describing Correlated Systems:
Density Functional Theory (DFT) with local (on-site) Coulomb interactions, U – specifically, DFT+U and DFT+DMFT – has been highly successful in accounting for electron-electron correlations and in describing the electronic structure of correlated systems. These methods provide better descriptions of key experimental features, such as the band gap, optical properties, and photoemission spectra, particularly in transition metal oxides (TMOs), and have made significant contributions to the theoretical condensed matter physics. However, despite these successes, recent studies highlight the limitations of these local theories in understanding the physics of correlated systems, emphasizing the importance of incorporating nonlocal (inter-site) Coulomb interactions.
Although the role of inter-site Coulomb interactions on electronic structures has been intensively discussed within the model community, particularly in various two-dimensional (2D) lattices, they emphasize the crucial role of the interaction strength V in determining charge density wave phases and selectively stabilizing different superconducting order parameters. Despite extensive research on 2D lattices, the effects of V in three-dimensional lattices, which more closely resemble real material systems, remain largely unexplored. Some methods, such as GW+DMFT, offer promising practical applications; however, their computation cost has limited their widespread use in the study of diverse materials.
Coulomb Interactions and Screening:
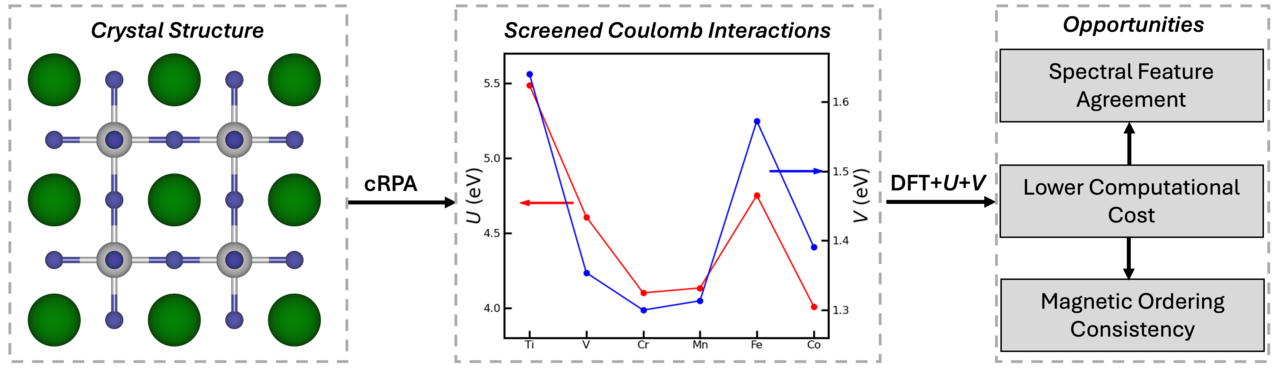
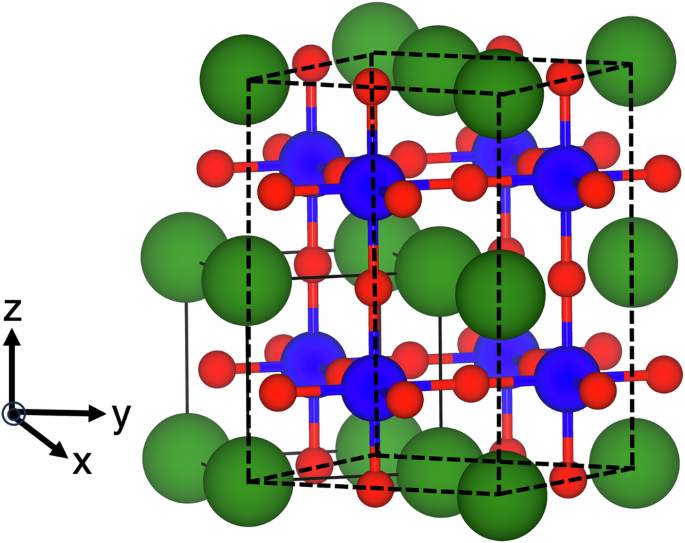
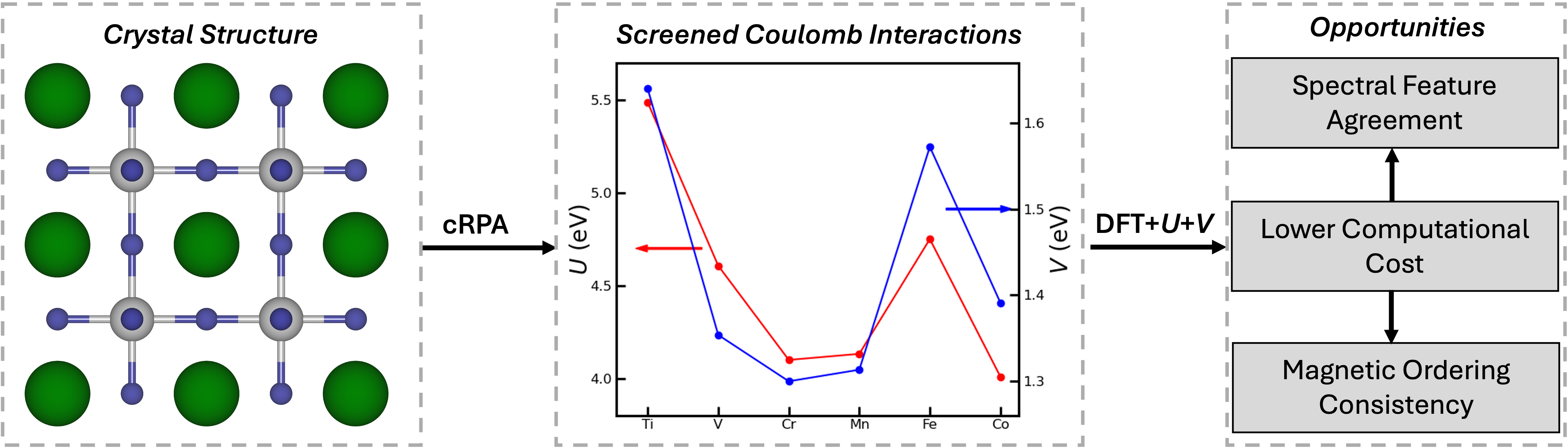
We have quantified the local and nonlocal Coulomb interactions using the constrained random phase approximation (cRPA) method. One of the useful characteristics of cRPA is its ability to identify the detailed screening channels of specific manifolds. The screening interactions are then quantified for each orbital using Wannier functions, accounting for both on-site and inter-site Coulomb interactions, as well as Hund's interaction parameters. In our work published in npj Computational Materials, we focused on representative transition metal oxides in the form of SrMO3 (M = 3d) perovskites (see Figure 1) and investigated both local and nonlocal interaction parameters.
Our study shows that, unlike the anticipated increase in Coulomb interaction with increasing in electron localization with higher electron filling, the competition between electron localization and screening from hybridized orbitals – primarily due to electronic screening from the d-d and d-p channels – determines the strength of screened Coulomb interactions. This results in the nonmonotonic behavior of both on-site and inter-site screened Coulomb interactions, as shown in Figure 1. We further highlight that nonlocal screening extends over interatomic distances, clearly demonstrating the significant strength of the inter-site interaction V, which is approximately 37% of the local interaction U. Additionally, we emphasize the importance of d-p screening, particularly in later TMOs, where pd-hybridization is strong.
Influence of Nonlocal Coulomb Interactions on Electronic Structure:
Nonlocal Coulomb interactions renormalize the bandwidth in a way that contrasts with the effect of on-site interactions. These effects have been extensively studied in extended Hubbard model studies. The inclusion of V leads to a widening of the d-bandwidth, particularly in systems with lower electron fillings, as predicted by model studies. This widening enhances the overall pd-hybridization, which in turn alters the bonding and anti-bonding levels. Specifically, for SrVO3, the shift in the t2g and eg peaks is in good agreement with experimental spectroscopic data. Here, we achieve these spectral features at a much lower computational cost using DFT+U+V rather than through GW+DMFT calculations, which demands higher resources.
Furthermore, at higher energy regimes, the inclusion of V leads to an overall pushing-up of the energy levels, demonstrating the multifaceted role of V in real systems. Unlike the cases with lower electron filling, the study revealed that the pd-hybridization in later TMOs causes the system’s behavior to deviate from the predictions of the idealized extended Hubbard model. Moreover, in later TMOs, the spectral weight at the Fermi level shifts to both higher and lower energy regions, a behavior akin to spectral weight transfer in the many-body perspective. Finally, our study demonstrates that the inclusion of inter-site Coulomb interactions (V) is essential for accurately reproducing the experimental magnetic order in these systems.
We believe our work provides valuable insights for applying first-principles theory with extended Coulomb interactions to real material systems within the strongly correlated physics community.
For further information, please refer to our published article: https://doi.org/10.1038/s41524-024-01454-9
Follow the Topic
-
npj Computational Materials
This journal publishes high-quality research papers that apply computational approaches for the design of new materials, and for enhancing our understanding of existing ones.

Related Collections
With Collections, you can get published faster and increase your visibility.
Recent Advances in Active Matter
Publishing Model: Open Access
Deadline: Sep 01, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in