Predicting & synthesizing ultra-hard materials
Published in Materials and Computational Sciences
In the ever-evolving landscape of materials science, the quest for novel materials with enhanced properties is a relentless endeavor. Recently, an interdisciplinary team of researchers has made a significant breakthrough in this field by employing a hybrid approach combining graph neural networks (GNNs) and density functional theory (DFT) to discover chemically modified higher tungsten borides. This innovative methodology not only accelerates the discovery process but also enhances the accuracy of predicting thermodynamic properties, paving the way for the synthesis of materials with superior mechanical characteristics.
The Challenge of Materials Discovery
The traditional path to discovering new materials often involves extensive experimental trials, which can be both time-consuming and resource-intensive. As computational methods and artificial intelligence (AI) advance, they offer promising alternatives for predicting crystal structures and their properties. However, one of the major challenges lies in improving material properties through alloying or doping, which requires navigating vast composition/configuration spaces efficiently.
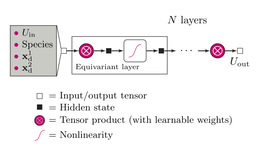
In this context, machine learning emerges as a powerful tool, particularly when applied to complex systems where the number of possible structure realizations is astronomically large. Recent advancements in geometric graph neural networks (GNNs) have opened new avenues for implementing transfer learning routines, enabling predictions based on small datasets derived from established databases like the Materials Project and AFLOWLib.
The Hybrid Approach: Combining GNNs and DFT
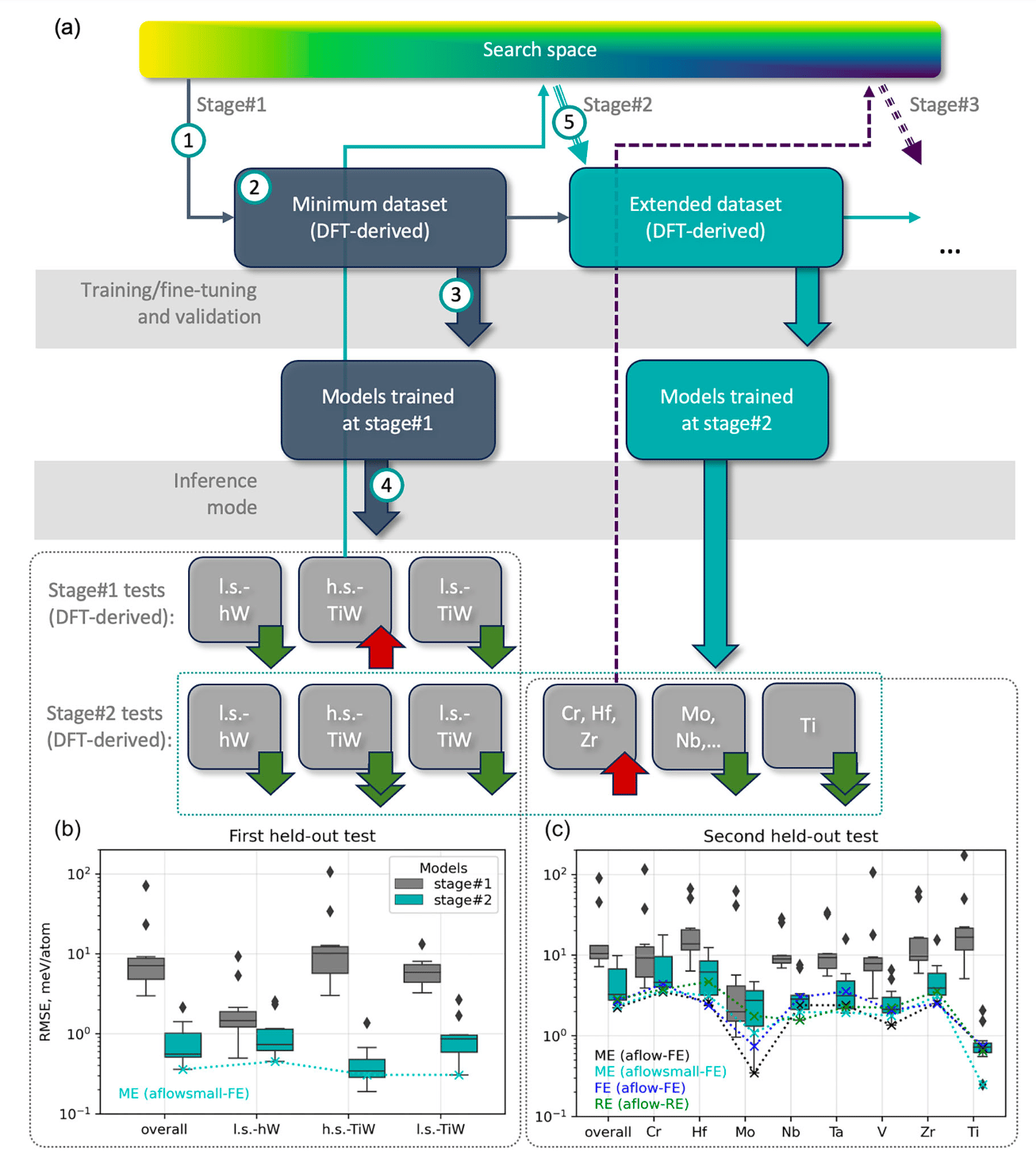
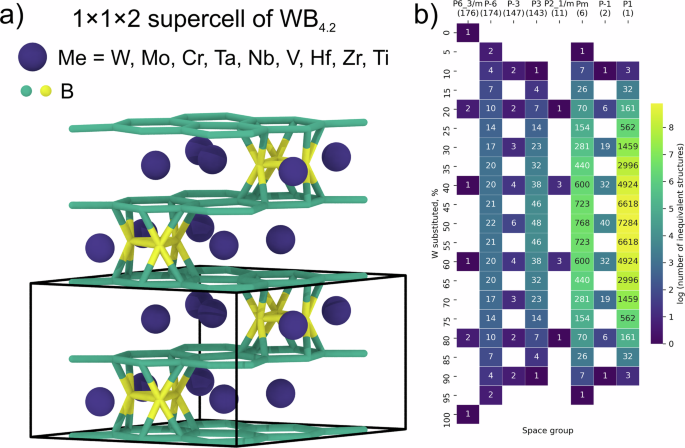
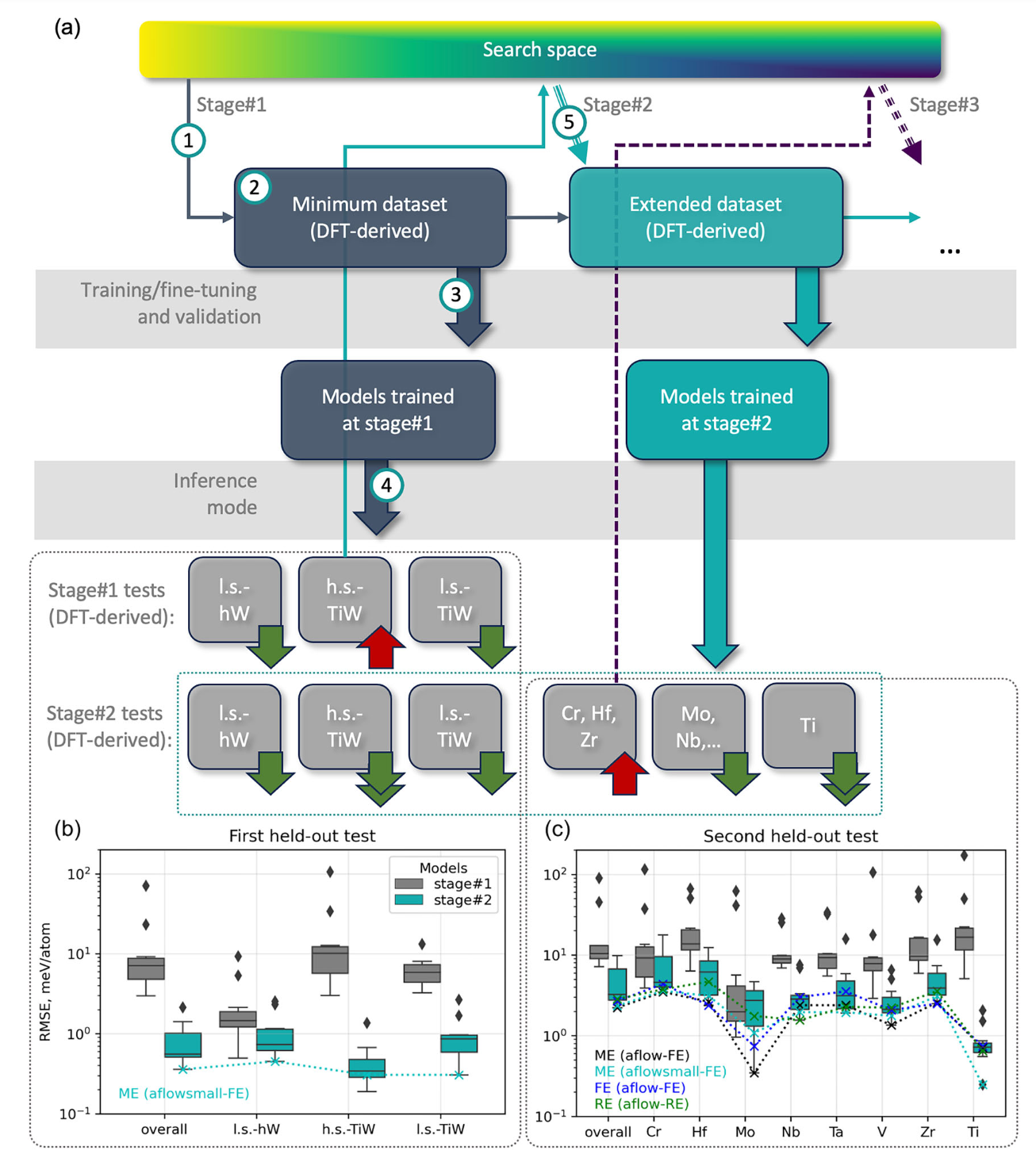
The research team focused on the higher tungsten boride, WB₄.₂, and explored eight metals as potential substitutes for tungsten. By constructing a search space comprising over 375,000 inequivalent crystal structures for solid solutions, they aimed to predict thermodynamic properties using GNNs fine-tuned on DFT-derived data. The goal was to identify the most promising candidates for chemical modification that could lead to enhanced mechanical properties.
Among the substituents considered, tantalum (Ta) emerged as the most favorable option, offering the widest range of predicted stable concentrations and significant improvements in mechanical properties. To validate these theoretical predictions, the researchers synthesized higher tungsten borides with varying Ta concentrations using a vacuumless arc plasma method. Vickers hardness measurements of WB₅₋ₓ samples with different Ta contents revealed a notable increase in hardness, confirming the efficacy of the proposed approach.
Methodology and Workflow
The workflow developed in this study involved several key stages:
-
Data Collection and Preparation : Starting with a 1 × 1 × 2 supercell of the WB₄.₂ crystal structure, the team generated a comprehensive set of symmetrically inequivalent substituent arrangements using the Supercell program. This step was crucial for building a robust dataset that captured the complexity of the system.
-
Model Training and Validation : Utilizing the Allegro architecture, the researchers trained GNN models on DFT-derived properties of approximately 200 entries. These models were then tested on "out-of-domain" entries to evaluate their predictive capabilities. The iterative process allowed for the refinement of the training dataset, ensuring high accuracy in predictions.
-
High-Throughput Screening : After validating the models, the team conducted high-throughput screening for stable compositions. This stage involved DFT calculations of mechanical properties for the predicted structures, providing insights into their stability and performance.
-
Experimental Synthesis and Characterization : Guided by theoretical predictions, the researchers synthesized powders and ceramic samples of W-Ta substituted higher tungsten borides. Advanced characterization techniques, including X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and Vickers microhardness measurements, were employed to assess the structural and mechanical properties of the synthesized materials.
Results and Implications
The results of this study are profound. The hybrid GNN/DFT approach successfully identified Ta as the optimal substituent for enhancing the mechanical properties of higher tungsten borides. Experimental validation confirmed that increasing Ta content led to a significant rise in Vickers hardness, highlighting the practical implications of the findings.
Moreover, the study underscores the importance of incorporating domain-specific knowledge during the prediction phase. By leveraging symmetry information and focusing on high-symmetry configurations, the researchers minimized randomness and improved the reliability of predictions. This approach not only enhances the efficiency of the discovery process but also reduces the risk of false positives, a common issue in high-throughput studies.
Future Perspectives
This work represents a paradigm shift in materials discovery, demonstrating how AI-driven methodologies can complement traditional experimental approaches. The integration of GNNs and DFT provides a scalable framework for exploring other families of functional materials, potentially leading to the development of new compounds with tailored properties.
As computational resources continue to evolve, the application of such hybrid approaches will become increasingly feasible across various domains of materials science. Furthermore, the ability to rapidly synthesize and characterize materials based on theoretical predictions opens new possibilities for accelerating innovation in fields ranging from electronics to energy storage.
In conclusion, the successful implementation of this hybrid strategy marks a significant milestone in the journey toward automated and physically informed materials discovery. By bridging the gap between theoretical predictions and experimental validation, this study sets a precedent for future research endeavors aimed at harnessing the power of AI to revolutionize materials design and development. 🌍
Good partnership (Skolkovo Institute of Science and Technology, AIRI, National Research Tomsk Polytechnic University, Sber) has resulted into good results!PhD in Computational Physics, Head of R&D at Sber AI, Scientific consultant at AIRI. Alma mater: MIPT 2019, ESPCI 2014, NSU 2011.
Follow the Topic
-
npj Computational Materials
This journal publishes high-quality research papers that apply computational approaches for the design of new materials, and for enhancing our understanding of existing ones.

Related Collections
With Collections, you can get published faster and increase your visibility.
Altermagnetic Materials: Theory, Simulation, and Renewed Perspectives
Publishing Model: Open Access
Deadline: Feb 28, 2027
Recent Advances in Active Matter
Publishing Model: Open Access
Deadline: Sep 01, 2026