Machine learning Hubbard parameters with equivariant neural networks
Published in Materials, Physics, and Computational Sciences
Scientists at EPFL and the Paul Scherrer Institute have demonstrated that machine learning can significantly reduce the time and computational cost of density-functional theory with extended Hubbard functionals (DFT + U + V). This widely used method enables the accurate simulation of complex materials containing transition-metal or rare-earth elements by mitigating self-interaction errors inherent in semi-local functionals, particularly in systems with partially-filled d and f electronic states.
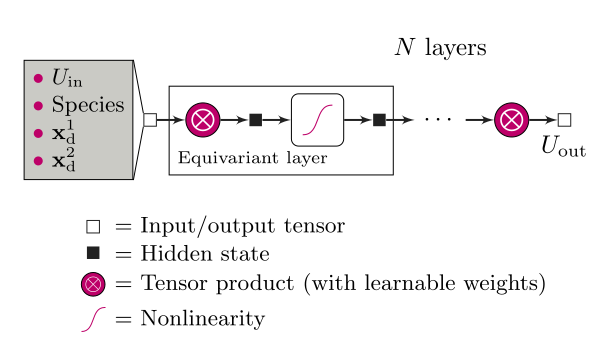
Achieving high accuracy with DFT + U + V depends on the precise determination of the on-site U and inter-site V Hubbard parameters. Traditionally, these parameters are either obtained through semi-empirical tuning—requiring prior knowledge—or through predictive but computationally expensive first-principles calculations. To address this challenge, the research team leveraged a recently developed class of neural networks called equivariant neural networks (ENNs), which offer a novel approach to learning and predicting Hubbard parameters with minimal computational overhead.
A New Machine Learning Approach
The team's machine learning model employs atomic occupation matrices as descriptors. These matrices directly capture the electronic structure, local chemical environment, and oxidation states of the system under investigation. The model is trained to predict Hubbard parameters computed self-consistently using iterative linear-response calculations within density-functional perturbation theory (DFPT) and structural relaxations.
The researchers trained two separate models—one for the U parameter and another for the V parameter—allowing them to work independently of one another. Using a dataset of 12 materials with varying crystal structures and compositions, the models achieved impressive results, with mean absolute relative errors of just 3% for U and 5% for V. Notably, these models also performed well in predicting downstream properties such as magnetic moments and voltages, demonstrating their robustness and practical applicability.
Key Advantages of the Machine Learning Model
The new approach replaces computationally demanding DFPT calculations while maintaining high accuracy for most practical applications. The model incorporates three crucial elements:
-
Atomic occupation matrices within the DFT + U + V framework, which serve as descriptors of the material’s geometry, oxidation states, and local chemical environment.
-
DFPT-based Hubbard parameters, which act as training targets.
-
Interatomic distances, which provide additional structural information.
By utilizing equivariant neural networks, the model leverages the inherent O(3) group structure of the occupation matrices, ensuring excellent performance even with scarce training data. This guarantees both high accuracy and strong transferability, making the model suitable for diverse materials with ionic, covalent, and mixed ionic-covalent interactions.
Implications for Materials Science
By circumventing computationally expensive self-consistent DFT or DFPT calculations, this machine learning model accelerates the prediction of Hubbard parameters with negligible computational cost. Given its robust transferability, the model has significant implications for materials discovery and design, enabling high-throughput calculations across various technological applications, including energy storage and quantum materials.
This study represents a significant step forward in integrating machine learning and electronic structure theory, providing an efficient and accurate alternative to traditional methods. Equivariant neural networks have already demonstrated state-of-the-art accuracy and transferability in machine learning interaction potentials, and this work is the first to incorporate electronic-structure degrees of freedom as explicit features in solids.
Explore More
Interested in the details? Read our full paper in npj Computational Materials: https://www.nature.com/articles/s41524-024-01501-5
Additionally, check out the NCCR-MARVEL highlight, where the story behind this paper is featured: https://nccr-marvel.ch/highlights/machine-learning-hubbard
Let’s accelerate materials science together!
Follow the Topic
-
npj Computational Materials
This journal publishes high-quality research papers that apply computational approaches for the design of new materials, and for enhancing our understanding of existing ones.

Related Collections
With Collections, you can get published faster and increase your visibility.
Altermagnetic Materials: Theory, Simulation, and Renewed Perspectives
Publishing Model: Open Access
Deadline: Feb 28, 2027
Recent Advances in Active Matter
Publishing Model: Open Access
Deadline: Sep 01, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in