Benchmarking machine learning models for predicting lithium ion migration
Published in Materials and Computational Sciences
In the ever-accelerating race to improve energy storage technologies, solid-state batteries have emerged as a promising frontier. These next-generation systems offer the potential for higher energy density, improved safety, and longer lifespans compared to traditional lithium-ion batteries. Central to this advancement is the development of fast ionic conductors—materials that allow lithium ions to move efficiently through a crystal lattice. However, identifying these materials has traditionally been a costly and time-consuming endeavor, reliant on high-throughput density functional theory (DFT) calculations. Now, a groundbreaking study published in npj Computational Materials offers a compelling solution: leveraging machine learning (ML) to accelerate discovery while maintaining accuracy.
The Challenge of Ionic Conductivity
Lithium ion transport in solid electrolytes is governed by migration barriers—the energy required for an ion to traverse between stable sites in the crystal lattice. Lower barriers mean faster ion movement, which translates into better battery performance. Historically, computational methods like DFT-based nudged elastic band (NEB) and ab initio molecular dynamics (AIMD) simulations have been used to calculate these barriers. While powerful, these techniques are computationally intensive, often requiring significant time and resources to explore large chemical spaces.
This bottleneck has driven researchers to explore alternative approaches, including simplified models such as the "pinball" model, empirical force fields like the Bond Valence Site Energy (BVSE) method, and more recently, ML-driven interatomic potentials. Yet, until now, no comprehensive benchmark existed to evaluate whether ML could reliably predict lithium ion migration barriers with accuracy comparable to DFT.
Introducing LiTraj: A Comprehensive Dataset for Machine Learning
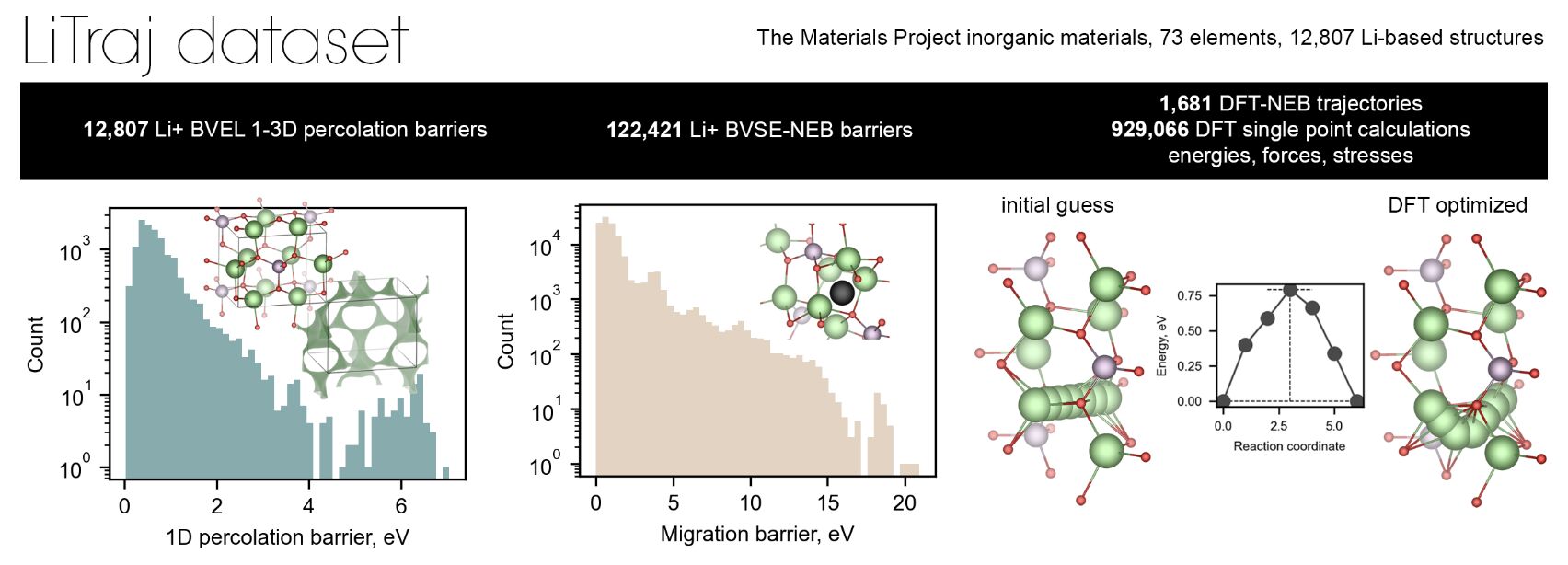
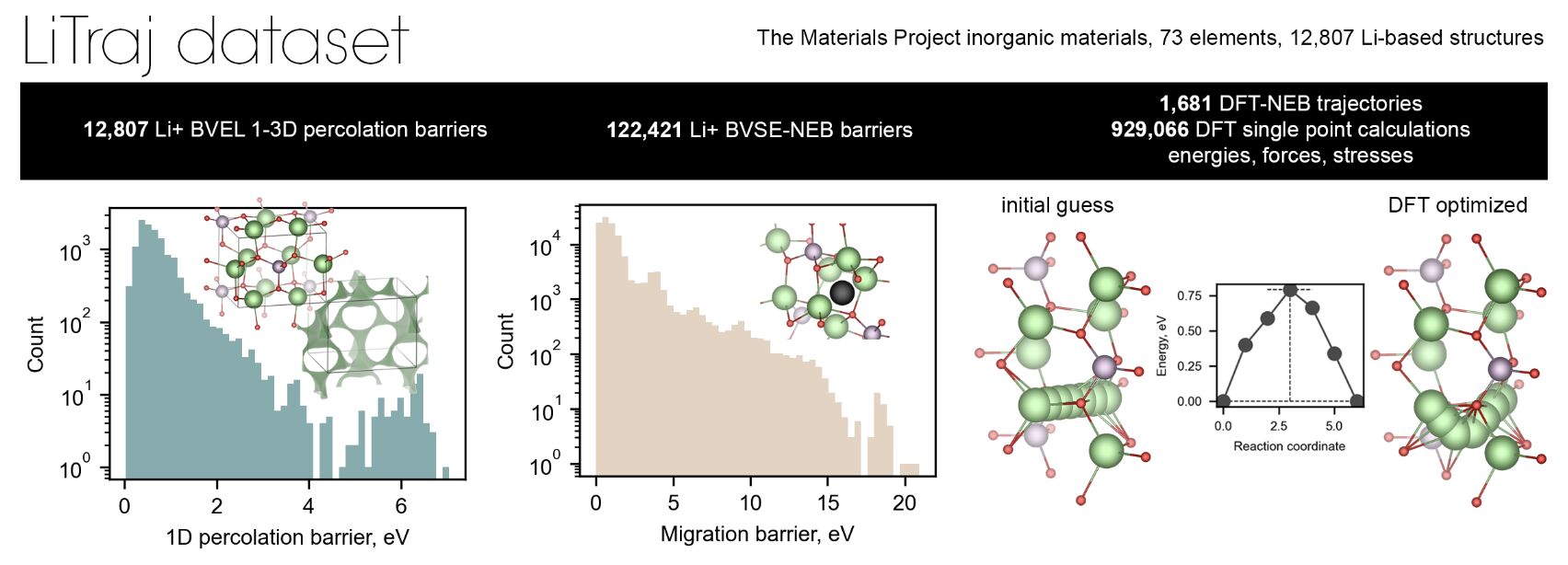
The authors of this study address this gap head-on by introducing LiTraj , a dataset comprising over 130,000 calculated migration barriers and trajectories across diverse crystal structures. This dataset includes:
- BVEL13k : Percolation barriers calculated using the BVSE method.
- nebBVSE122k : Migration barriers derived from BVSE-NEB optimization.
- nebDFT2k : High-fidelity migration barriers calculated via DFT-NEB.
- MPLiTrj : Over 900,000 atomic configurations capturing energies, forces, and stress tensors during NEB optimization.
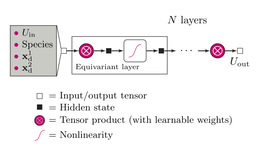
With LiTraj, the researchers benchmarked both classical ML models and graph neural networks (GNNs), evaluating their ability to distinguish between fast and poor ionic conductors. Moreover, they tested universal ML interatomic potentials (uMLIPs)—models capable of predicting not only scalar properties but also optimal migration pathways and corresponding energetics.
Machine Learning Models Rise to the Challenge
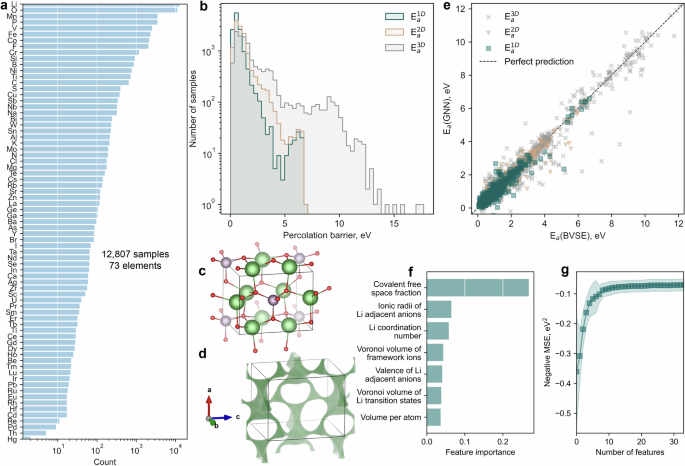
The results were striking. Classical ML models like random forest (RF), support vector regression (SVR), and gradient boosting (XGBRF), when trained on carefully engineered features based on Voronoi tessellations and elemental characteristics, demonstrated reasonable accuracy in predicting percolation barriers. However, GNNs—particularly M3GNet, Nequip, and Allegro—outperformed classical models, especially for higher-dimensional percolation maps.
For example, M3GNet achieved a root mean square error (RMSE) of just 0.2–0.5 eV across 1D, 2D, and 3D percolation barriers. Feature importance analysis revealed that descriptors such as covalent free space, Voronoi volumes around Li sites, and coordination numbers played crucial roles in prediction accuracy.
When it came to migration barrier prediction, transfer learning strategies further enhanced model performance. By pre-training on BVSE-derived data and fine-tuning on DFT-derived barriers, the researchers showed that hybrid approaches can bridge the gap between speed and accuracy.
Universal ML Interatomic Potentials: Predicting Pathways and Energetics
Perhaps the most exciting aspect of the study was the evaluation of uMLIPs—machine learning models designed to replicate quantum mechanical interactions at a fraction of the computational cost. Four state-of-the-art uMLIPs—M3GNet, CHGNet, SevenNet, and MACE—were tested against DFT-NEB data for their ability to reproduce optimal Li+ migration paths and associated energy profiles.
Among them, SevenNet and MACE stood out, achieving mean absolute errors (MAEs) as low as 0.07–0.08 eV for systems without transition metals (TMs). For TM-containing systems, where DFT typically requires Hubbard U corrections and spin polarization, errors were higher (~0.20–0.21 eV). However, fine-tuning these models on LiTraj data significantly improved their accuracy, reducing MAEs to ~0.10–0.11 eV and demonstrating that uMLIPs can be adapted to specific computational schemes.
Notably, the path-averaged MAE (PA-MAE) metric—a measure of how closely predicted migration pathways match DFT-derived ones—revealed that uMLIPs could capture structural details of ion diffusion with remarkable fidelity. This opens new possibilities for exploring bottlenecks in ion transport and guiding materials design.
Real-World Application: Screening Protective Coatings for Solid-State Batteries
To demonstrate the practical utility of their approach, the researchers applied the fine-tuned SevenNet model to screen protective coatings for stabilizing the interface between Li10GeP2S12 (LGPS) solid electrolyte and LiCoO2 cathode material—an important challenge in all-solid-state battery design. Using criteria such as electrochemical stability and interface compatibility, they identified several promising candidates, including known conductors like Li3AlF6 and Li3PO4, as well as novel materials like LiYF4 and Li2ZnCl4.
These findings highlight how ML-enhanced workflows can accelerate materials discovery, offering a viable alternative to traditional high-throughput screening pipelines.
Limitations and the Road Ahead
Despite its successes, the study acknowledges several limitations. First, the training data is biased toward near-equilibrium structures due to the use of BVSE-NEB as an initial guess for DFT-NEB calculations. Second, the effectiveness of fine-tuning protocols—such as choice of loss function, optimizer, and hyperparameters—remains underexplored. Finally, the current focus on vacancy-mediated Li+ migration does not account for other important mechanisms, such as interstitial or concerted migration, which may dominate in superionic conductors.
Future work should aim to expand the dataset to include these phenomena, refine fine-tuning strategies, and explore how different exchange-correlation functionals affect barrier predictions. Moreover, integrating AIMD-derived conductivity data into ML models could provide a more holistic understanding of ionic transport.
Conclusion
This study represents a major step forward in applying machine learning to materials science, particularly in the context of fast ionic conductors. With the release of the LiTraj dataset and the demonstration of ML's predictive power, researchers now have a powerful toolkit to accelerate the discovery of next-generation solid electrolytes.
PhD in Computational Physics, Head of R&D at Sber AI, Scientific consultant at AIRI. Alma mater: MIPT 2019, ESPCI 2014, NSU 2011.
Follow the Topic
-
npj Computational Materials
This journal publishes high-quality research papers that apply computational approaches for the design of new materials, and for enhancing our understanding of existing ones.

Related Collections
With Collections, you can get published faster and increase your visibility.
Altermagnetic Materials: Theory, Simulation, and Renewed Perspectives
Publishing Model: Open Access
Deadline: Feb 28, 2027
Recent Advances in Active Matter
Publishing Model: Open Access
Deadline: Sep 01, 2026