Genomic footprints of activated telomere maintenance mechanisms in cancer
Published in Cancer


Cancer cells are immortal, a property that they share with stem cells. This is achieved by constant elongation of their telomeres, which are the structures that protect the end of linear chromosomes and that are shortened each time the cell divides. And indeed, about ~ 85% of all tumors reactivate the canonical telomere maintenance that is active in stem cells, but downregulated in most somatic tissues. The rate limiting step in this biological process is the expression level of the gene TERT. Its upregulation in different tumor types is a prime example of convergent cancer evolution, as it is achieved through a multitude of aberrations including gene amplification, enhancer hijacking, recurrent promoter mutations, epigenetic alteration of repressor elements or the activation of transcription factors such as MYC and AR. The remaining 15 % of tumors rely on alternative lengthening of telomeres (ALT), which comprises all mechanisms that elongate telomeres without TERT and the telomerase complex. ALT is surprisingly understudied, but current models imply recombination-based mechanisms. It has been described before that this process alters the composition of the telomeres from ~10-12 kbp long repetitions of the TTAGGG repeat hexamer into more complex patterns.
When the call for projects of the Pan-cancer Analysis of Whole Genomes (PCAWG) study reached us about six and a half years ago, we considered what type of question could be answered with a set of more than 2,500 pairs of matched cancer and control genomes that was not addressable before. As we had recently found that WGS data was a powerful tool to detect ALT-positive glioblastomas and medulloblastomas, we considered a detailed characterization of the genomic footprints of telomere maintenance mechanisms. At that time the literature on this phenomenon was very sparse and results were mainly derived from cancer cell lines rather than primary tumor samples. Most prominently, no larger dataset of sequenced ALT rearranged telomeres was available.
Considering that about 3 out of 20 patients worldwide suffer from ALT-positive tumors, which would stop dividing after a few dozen cell generations if the ALT mechanism could be effectively blocked, this appeared to be a missed opportunity for the development of new treatment options. All the more as ALT is a process, which is exclusively active in cancer and has no known function in healthy human cells. In consequence, any treatment that targets ALT directly bears the promise of having only mild side effects. Furthermore, the clinical trajectory of TERT-positive and ALT-positive cancers can differ dramatically, where in some cancers the one type of telomere maintenance mechanism (TMM) marks a more aggressive phenotype, while the roles are reversed in another. Therefore, a cost-efficient diagnosis of the active TMM directly from sequencing data is also of high clinical relevance.
After submission of our research proposal to the PCAWG steering committee, the first step towards an in silico telomere analysis was the development of a software that reliably identifies all reads with a high content of telomeric sequence from next-generation sequencing (NGS) datasets. Here, we noted that the existing tools already established good criteria, but usually operated on unaligned data. This had two drawbacks: Firstly, pseudo-telomeric sequences from intra-chromosomal loci are included into the analysis and thus add a constant bias to all results. Secondly, the integration of telomeric sequence into the cancer genome and the capping of genomic double-strand breaks by telomeric sequence, the so called telomeric healing, are indistinguishable from intra-telomeric signals. To this end, we designed the TelomereHunter software, and benchmarked it on different datasets and sequencing protocols together with our partners from the CancerTelSys consortium, the ICGC PedBrain project and the NCT MASTER program. In this the research environment at the German Cancer Research Center (DKFZ) proved to be a critical catalyst, as it is probably one of the few places world-wide in which you find a comparable concentration of expertise on Cancer Telomere biology, Cancer Genomics and Clinical Sequencing within 5 minutes of walking distance.
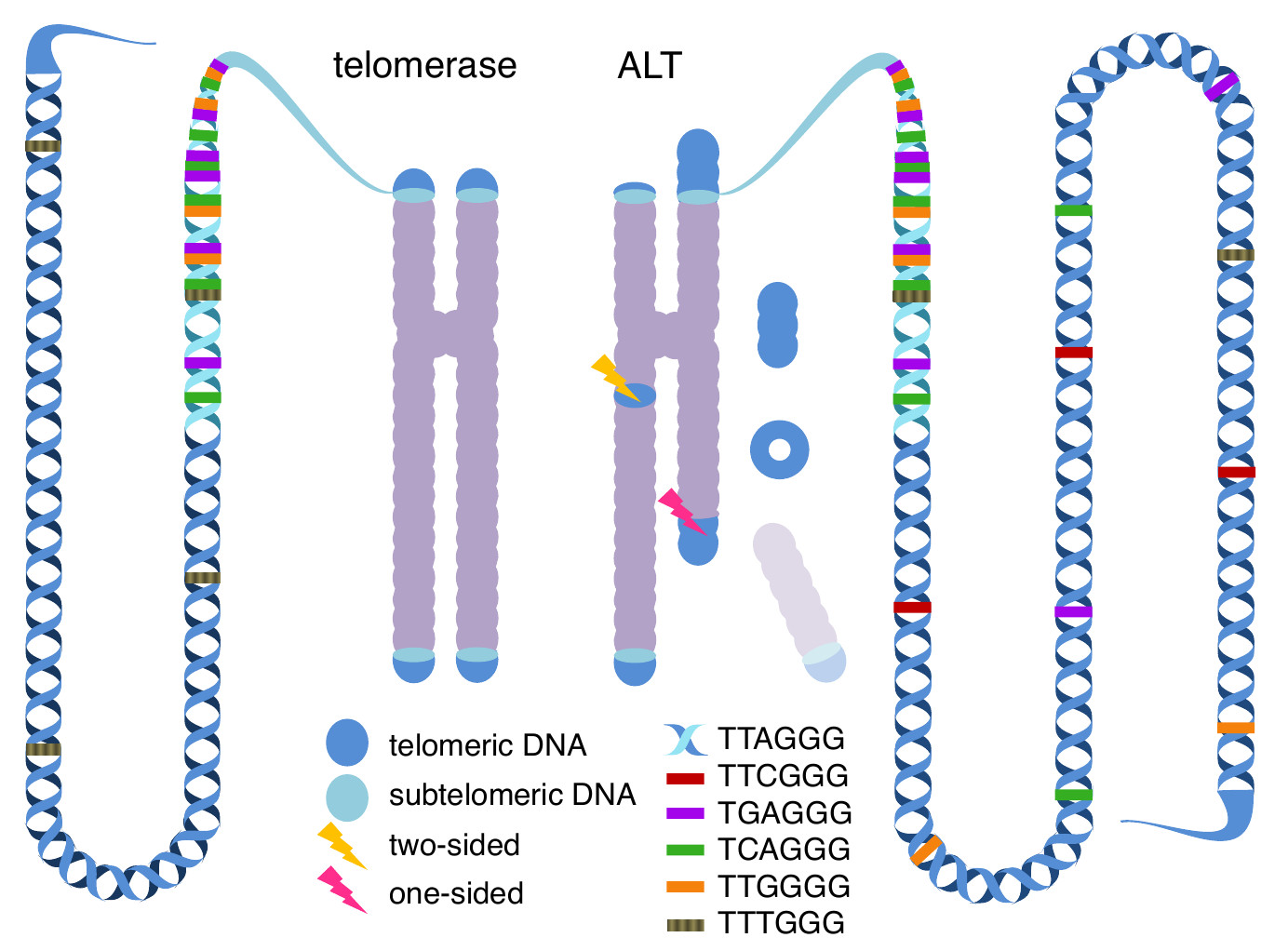
Having established a well characterized software, we started to profile the cancer genomes. Due to the repetitive nature of telomeres and the limitations of short-read sequencing, reconstruction of ALT-telomeres was not an option. Therefore, we focused on the analysis of the local sequence composition of telomeric reads. Building on earlier observations that variants of the classic TTAGGG repeats often conserve the triple guanine, we performed a first round of complexity reduction by counting all 64 variants of the NNNGGG pattern in our data. We soon discovered that only a minority of these telomeric variant repeats (TVRs) were correlated with the presence of ALT hallmark mutations in the genes ATRX and DAXX. Furthermore, we discovered that the neighbourhood of these TVRs was relevant, as TVRs embedded in unaltered telomeric sequence showed a stronger signal. Using these singleton TVRs, we also found that the hexamere TTTGGG is strongly depleted in ALT-positive tumors, a phenomenon that has not been described before. Finding an explanation for this observation may lead to a critical refinement of our understanding of the underlying molecular mechanisms.
Using the alignment information of intra-chromosomal reads and the matched control genomes, it was also possible to identify somatic de novo insertions of telomeric sequences. These often co-located with only one side of a genomic breakpoint, which can be interpreted as the healing of a double strand break by establishment of a new functional chromosome end. These events were remarkably enriched in ALT-positive cases, especially when the overall telomere content was high and microhomology to telomeric sequences existed. This indicates that the same DNA repair mechanisms, which are active during the recombination-based telomere elongation, are also active during telomere healing, and that the abundance of telomeric templates facilitates both processes. This may also explain why abundant extra-chromosomal telomeric DNA – in linear as well as in circular form - is observed in ALT positive patients. If the recombination is a stochastic process in which a break, a matching template and the DNA repair machinery have to assemble, then increasing the amount of templates has a positive impact on the kinetics.
Training a machine learning algorithm on all these features yielded a classifier that was able to predict ALT positive tumors with high specificity, showing that beside the pure number of genomic breakpoints two of the newly described singleton TVRs were the most predictive features. Unsupervised analysis of an integration of the telomere features with the data from the other working groups for the Replicative immortality chapter of the PCAWG marker paper then discovered four distinct types of TMMs of which two were TERT-based and two were ALT-based.
The translational impact of our study was immediate, as we formed a cooperation with the INFORM precision oncology program. The focus of this program is on paediatric cancers, which show a particular enrichment of ALT cases. Since 2018 TelomereHunter is applied routinely to the NGS data produced in this program to prioritize samples for targeted validation of the ALT status. This information is then provided for the molecular tumor board to allow for a more informed treatment proposal. Currently, ALT specific therapy options are still limited, but the molecular data that the community has accumulated in the last six years may provide a starting point to change this in the future.
Follow the Topic
-
Nature Communications

An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Advances in neurodegenerative diseases
Publishing Model: Hybrid
Deadline: Mar 24, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in