GRID1/GluD1 homozygous variants linked to intellectual disability and spastic paraplegia impair mGlu1/5 receptor signaling and excitatory synapses
Published in Neuroscience and Biomedical Research
Background
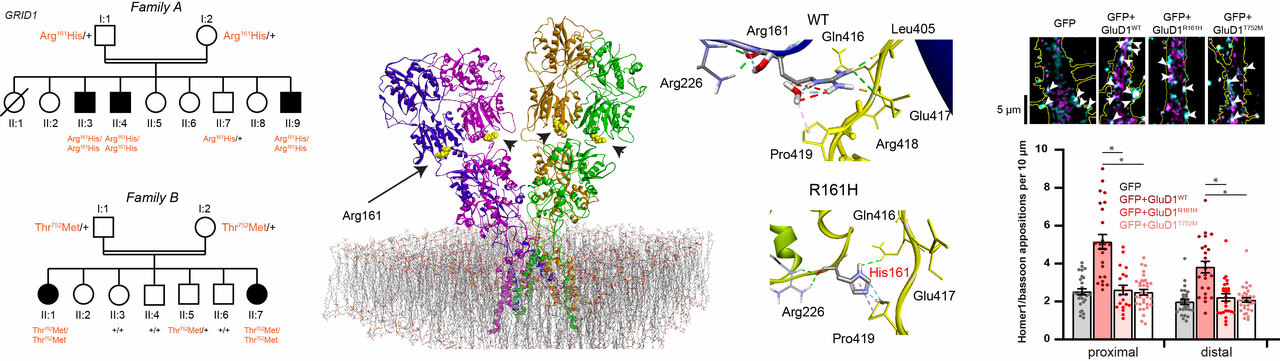
Intellectual disability (ID) and spastic paraplegia (SPG) are clinically and genetically heterogeneous central nervous system disorders that are frequently associated. Conversely, the triple combination of ID, SPG and glaucoma is a rare syndrome whose genetic basis is unknown.
- The ionotropic glutamate delta receptor GluD1, encoded by the GRID1 gene, is involved in synapse formation, function, and plasticity. GluD1 does not bind glutamate, but instead cerebellin and D-serine, which allow the formation of trans-synaptic bridges, and trigger transmembrane signaling.
- GluD1 is engaged in a functional crosstalk with Gq-coupled metabotropic glutamate receptors (mGlu1/5)
- Despite wide expression in the nervous system, pathogenic GRID1 variants have not been characterized in humans so far.
Main Results
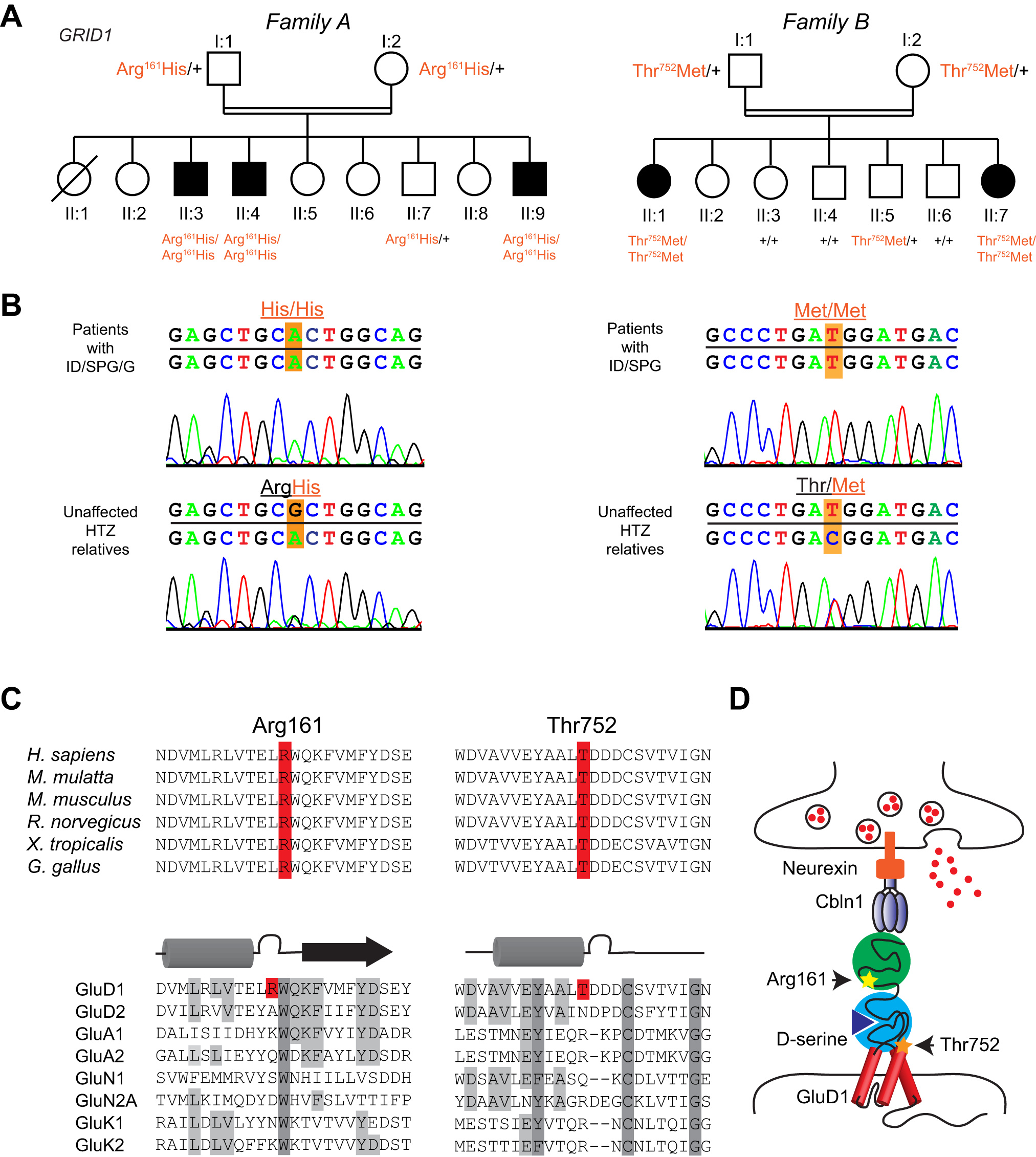
- We found homozygous missense GRID1 variants in patients from two unrelated consanguineous families presenting with intellectual disability and spastic paraplegia, without (p.Thr752Met) or with (p.Arg161His) glaucoma.
- The Arg161His and Thr752Met mutations are localized in the extracellular cerebellin and D-serine binding domains, respectively. Molecular modeling and experimental studies indicate that Arg161His alters the interactions between the cerebellin and D-serine binding domains but impacts neither cerebellin nor D-serine binding. The Thr752Met mutation causes a stiffening of the D-serine binding domain, which leads to decreased D-serine sensitivity but has no effect on cerebellin binding.
- Upon expression in dissociated mouse cortical neurons, we found that both GluD1 mutants hampered mGlu1/5-dependent Ca2+ increase and ERK activation, as compared to wild-type GluD1.
- Both mutations impaired the enhancing effect of GluD1 on excitatory synapse formation in mouse hippocampal dissociated neurons and organotypic slice cultures.
Conclusion
We characterize the first pathogenic variants of the GRID1 gene in patients presenting with ID and SPG with or without glaucoma and provide evidence that these variants impair mGlu1/5 signaling and synapse density. These findings unravel the importance of GluD1 receptor signaling in sensory, cognitive and motor functions of the human nervous system.
Follow the Topic
-
Molecular Psychiatry

This journal publishes work aimed at elucidating biological mechanisms underlying psychiatric disorders and their treatment, with emphasis on studies at the interface of pre-clinical and clinical research.
Your space to connect: The Psychedelics Hub
A new Communities’ space to connect, collaborate, and explore research on Psychotherapy, Clinical Psychology, and Neuroscience!
Continue reading announcement

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in