How B-cell lymphoma hijacks signals from the microenvironment to survive
Published in Biomedical Research
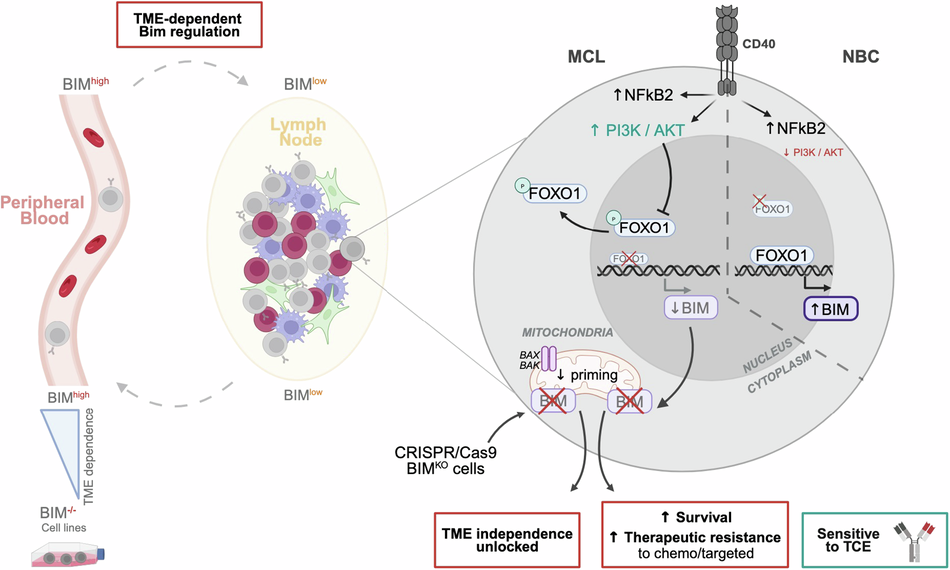
Both solid and hematological malignancies are now recognized as complex ecosystems. Indeed, tumor cells do not act alone: they are embedded within specific cellular and soluble environments, the tumor microenvironment (TME). Together with stromal cells, immune cells, and soluble factors, they shape a dynamic network that can promote survival and therapeutic resistance.
Mantle cell lymphoma (MCL), a rare but aggressive B-cell lymphoma, is no exception. Although it develops preferentially in lymph nodes (LN), tumor cells rapidly disseminate to peripheral blood (PB), bone marrow, and spleen. This multiple localization offers a unique opportunity: by comparing tumor cells across different microenvironmental niches, we can dissect how the microenvironment shapes tumor behavior.
Modeling the lymph node microenvironment outside the body
Back in 2015, our team set out to model the LN microenvironment ex vivo. We developed a PB primary cell co-culture system combining CD40L-expressing fibroblasts, mimicking key stromal and lymphoid interactions, with a cytokine cocktail representing soluble myeloid factors. Strikingly, this model reproduced nearly 70% of the transcriptional differences observed between circulating tumor cells and their LN-resident counterparts. Using this system, we demonstrated that MCL cells are highly dependent on immune-derived cues for survival and proliferation. This raised a central question: what molecular mechanisms hide behind this microenvironment-driven survival and can we target them to improve patient treatment?
Among the key molecular changes observed in LN, one finding stood out: a profound dysregulation of BCL2 family genes expression. Anti-apoptotic genes such as MCL1 and BCL-xL were upregulated, while pro-apoptotic genes, particularly BCL2L11 (the gene encoding BIM), were downregulated. At the time, our efforts focused on BCL-xL and its connection to NF-κB signaling and drug resistance, work that contributed to therapeutic combination strategies now used clinically; and BIM remained in the background, a discreet yet potentially decisive player.
The return of BIM
In 2019, we revisited BIM. The BCL-2-interacting mediator of cell death (BIM) is a critical pro-apoptotic effector in B-cell lymphomas. Its expression level and stability strongly influence therapeutic response, and accumulating evidence supports its central role in MCL pathophysiology. Low BIM expression in LN-resident tumor cells has been associated with poor prognosis. In murine models, deletion of BCL2L11 in the context of Cyclin D1 overexpression leads to an MCL-like disease. Moreover, homozygous deletion of BCL2L11 is found in approximately 50% of MCL cell lines.
Yet here lies a paradox: while BIM deletion is common in cell lines, it is rarely observed in primary patient samples. When we examined primary MCL cells, we confirmed the absence of genetic deletion. Surprisingly, circulating MCL cells expressed even higher levels of BIM than circulating normal B cells (NBC). Given BIM’s role in promoting apoptosis, this observation was unexpected. It suggested two important ideas. First, that loss of BIM likely provides a selective advantage during cell line immortalization. Second, in patients, MCL cells must rely on a non-genetic mechanism to suppress BIM and our previous work pointed directly to the LN microenvironment.
Intriguingly, we observed the opposite regulation in NBC: LN-resident NBC expressed higher BIM levels compared to their circulating counterparts. BIM repression was therefore tumor-specific and actively driven by microenvironmental signaling.
CD40L/PI3K/FOXO1 : a molecular switch
Using our TME co-culture system, we confirmed that LN-like conditions repress BIM at both transcriptional and protein levels in MCL cells, while inducing the opposite effect in NBC. Dissecting the individual components of the model revealed that BIM repression was driven specifically by CD40L stimulation.
We then searched for the transcription factors responsible for this differential effect. Computational analysis identified FOXO1, a known positive regulator of BIM expression. Immunofluorescence experiments revealed a striking divergence. In NBC, FOXO1 accumulated in the nucleus under co-culture conditions. In contrast, in MCL cells, CD40L stimulation activated the PI3K/AKT pathway, inducing phosphorylation of FOXO1 at serine 256, an event known to trigger its nuclear export and inactivation. Pharmacological inhibition of PI3Kδ using idelalisib restored BIM expression and prevented FOXO1 cytoplasmic relocation, directly demonstrating that CD40L-dependent BIM repression operates through the PI3K/AKT-FOXO1 axis.
What happens when BIM is lost?
To evaluate the functional consequences of BIM repression, we generated BIM knockout (KO) primary cells using CRISPR/Cas 9 technology. Primary MCL cells typically do not survive ex vivo without microenvironmental support. Remarkably, loss of BIM promoted their survival and immortalization, enabling the establishment of proliferating cell lines. This confirmed a direct link between BIM expression and TME dependence.
Using isogenic BIM KO cell lines, we uncovered several features of BIM-deficient cells:
(1) improved survival under stress conditions (serum-free culture), (2) increased tumorigenic capacity and aggressiveness (in ovo model) and (3) reduced mitochondrial priming. This last finding was particularly concerning. Indeed, reduced mitochondrial priming translated into broad resistance to therapies currently used in the clinic, including chemotherapy and targeted agents, most of which rely on the intrinsic apoptotic machinery.
But would BIM loss also compromise immunotherapy ? We tested T-cell engagers (TCEs), which redirect cytotoxic T lymphocytes toward tumor cells. In co-cultures with allogeneic T cells from healthy donors, cytotoxicity remained intact regardless of BIM expression levels, suggesting that TCE-mediated killing appeared independent of tumor cell BIM status.
A microenvironment-driven survival program
Taken together, our findings suggest that in MCL, the lymph node does not merely host the tumor, it teaches it how to survive. Through CD40L-mediated activation of the PI3K/AKT–FOXO1 pathway, it silences BIM, reduces mitochondrial priming, and reshapes treatment sensitivity.
Importantly, this mechanism is reversible. Targeting PI3K/AKT signaling may restore apoptotic competence, while immunotherapeutic strategies such as T-cell engagers may bypass intrinsic resistance mechanisms altogether.
Follow the Topic
-
Leukemia
This journal publishes high quality, peer reviewed research that covers all aspects of the research and treatment of leukemia and allied diseases. Topics of interest include oncogenes, growth factors, stem cells, leukemia genomics, cell cycle, signal transduction and molecular targets for therapy.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in