Linking mutations to cancer development
Published in Cancer
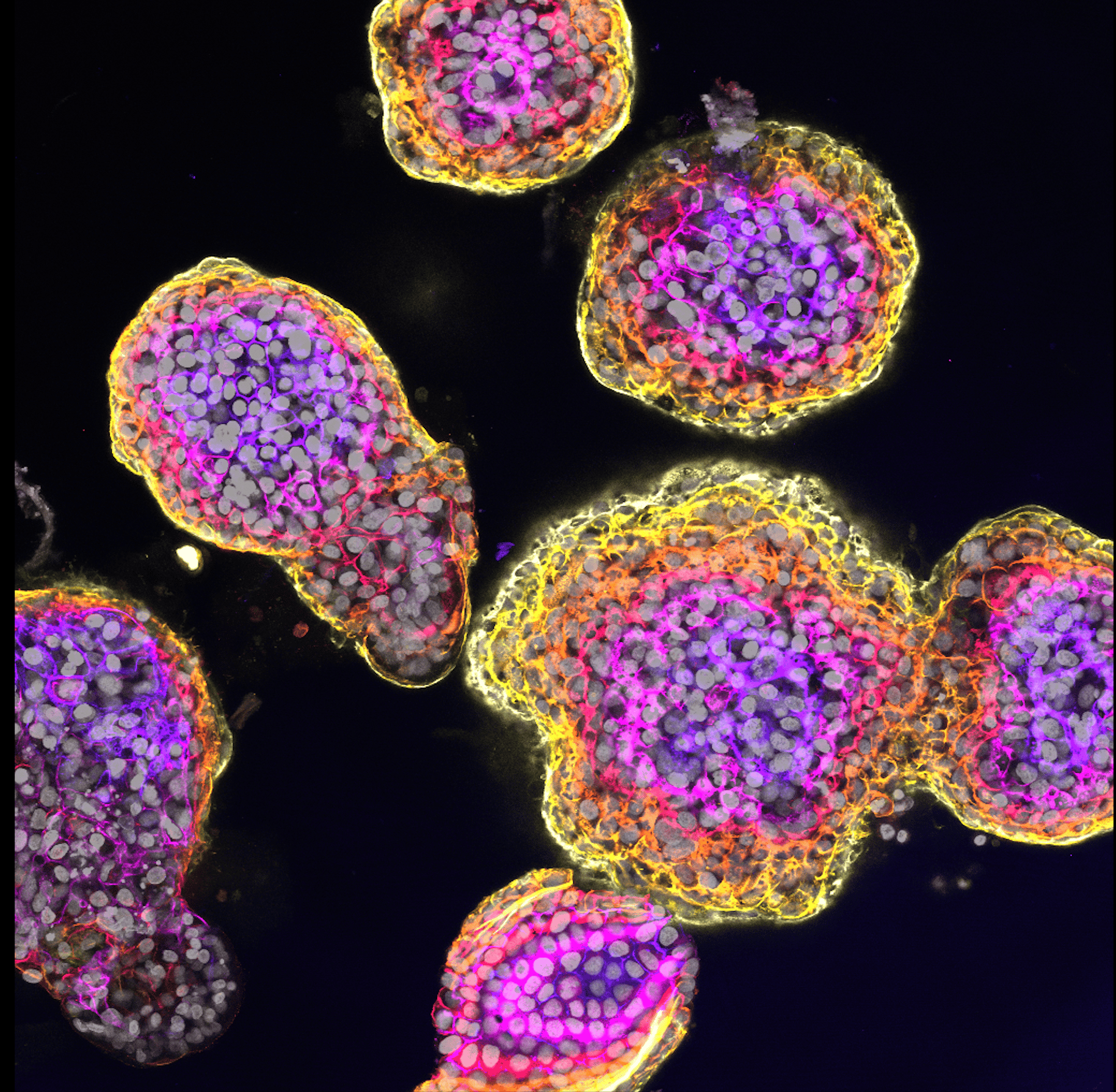

Genomic analyses of cancer tissues are vital to identify and link genetic aberrations to specific types of cancer. The next step is to try and understand how these mutations contribute to cancer development. Certain mutations will have more drastic effects and are necessary to drive tumorigenesis, others may be necessary in the process to further develop into a more aggressive tumor, while other mutations may be referred to as "passengers", i.e. they happen but are not essential for cancer development. On top of that, the order in which mutations occur is often not random and may be necessary to get the "right" tumor. A well-known example is the Vogelstein cascade of the most common subtype of colorectal cancer.
In order to provide relevant answers to the above problems, a model system ideally needs to meet two criteria: 1) it should be able to faithfully mimic the transformation of an initially healthy tissue into a transformed one and 2) it should be easily amenable to genetic modifications in order to screen for (combinations of) multiple mutations. The popularity of human organoids as a robust in vitro platform to address biological questions has immensely grown over the past years. Combining organoids with CRISPR gene editing tools establishes an extremely powerful platform to study cancer mutations. Two pioneering studies in 2015 clearly demonstrated this by modelling colorectal cancer in human intestinal organoids1,2.
A focus of our lab is devoted to study the liver in health and disease. Over the past years, we have shown that combining CRISPR with human liver organoids holds powerful tools to model cholangiocarcinoma3, study hepatocyte division and polyploidy4, and most recently to build models of non-alcoholic fatty liver disease5. Now, in our latest study6, we turned our attention to a specific type of liver cancer that sparked our interest due to its peculiar features: this cancer, fibrolamellar carcinoma (FLC), typically occurs in adolescents or young adults, and -in contrast to other types of liver cancer- develops in the absence of pre-existing disease or predisposing factors, such as alcohol, fatty liver, viral infections, etc.
The genetic background of FLC appears extremely "clean". In 2014, genomic efforts discovered a recurrent fusion event between the DNAJB1 gene and the PRKACA gene to establish a chimeric fusion transcript6, DNAJB1-PRKACAfus. The fusion has since been found in most of the classical FLC cases. More recently, a novel genetic background was linked to FLC-like tumors, which present with very similar histological features. These FLC-like tumors carry inactivating mutations in BAP1, often combined with PRKAR2A mutations7. Both PRKACA and PRKAR2A encode subunits of the protein kinase A (PKA), highlighting that genetic aberrations in PKA signaling components may be vital to develop FLC.
We set out to model these two different genetic FLC backgrounds in human hepatocyte organoids to understand the role and biological consequences of each specific FLC mutation6. To our surprise, we noted drastic differences between DNAJB1-PRKACAfus organoids versus BAP1KO;PRKAR2AKO organoids. On a histological level, the fusion-bearing organoids retained a phenotype that was more alike wild type organoids, while BAP1-mutated organoids appeared more transformed: they adopted a circular shape, turned dark, and displayed nuclear dysplasia.
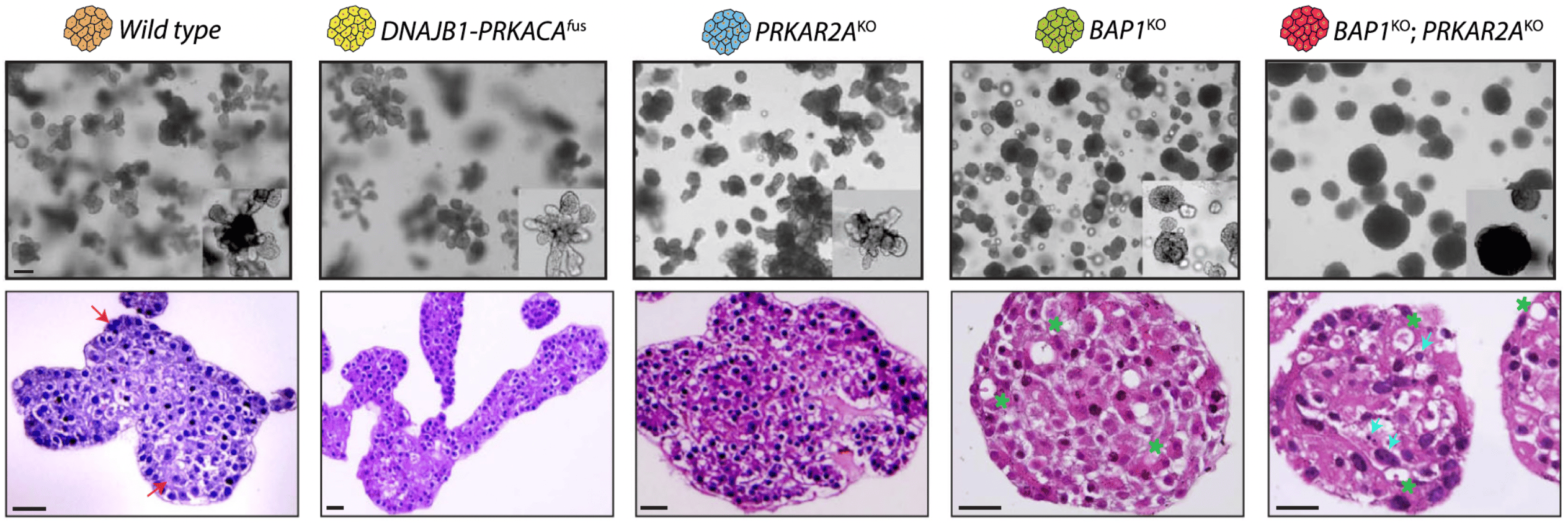
We performed a comprehensive characterization of their transcriptomes and protein marker expression. Intriguingly, all FLC mutations induced a certain degree of hepatocyte dedifferentiation, i.e. the cells started to lose some hepatocyte-specific marker expression upon introduction of either the fusion or BAP1-related mutations, a feature also observed in FLC tumor tissues. Yet, only the combined loss of both BAP1 and PRKAR2A led to hepatocyte transdifferentiation into ductal/progenitor-like cells. These double mutant cells had now lost many hepatocyte markers and started to express ductal cell markers. We also found that these cells could exclusively grow in a ductal cell environment and displayed cancer stemness features, demonstrating their gain of altered environmental sensitivity. We also performed FLC mutant-tumor comparisons. We noted that both DNAJB1-PRKACAfus organoids as well as the BAP1KO;PRKAR2AKO organoids shared similarities with their respective FLC tumor types. Again, a stronger correlation was found with the latter double mutant organoids.
Our study reveals important aspects about the biological impact of the different FLC mutations when introduced in human hepatocytes. Since FLC tumors share features of both hepatocytes and ductal cells, the cell-of-origin has been a debated topic. Our organoid data underscore that mutations can impose a cell identity switch, and thus that the cell identity features of a tumor do not always match their cellular origin, but could equally be the result of a cellular transdifferentiation event.
We are also left with novel questions that require more research: what is the reason for the relatively mild phenotype of DNAJB1-PRKACAfus organoids? Are there for example other tumor developing requirements that we momentarily miss? And what is the exact biological explanation for the cooperative loss of BAP1 and PRKAR2A in driving a more aggressive cancer phenotype? Our future work will be devoted to address these intriguing scientific questions.
References
1. Drost et al. Nature 521 43-47, 2015.
2. Matano et al. Nat Med 21 256-262, 2015.
3. Artegiani et al. Cell Stem Cell 24(6) 927-943, 2019.
4. Artegiani*, Hendriks* et al. Nat Cell Biol 22(3) 321-331, 2021.
5. Hendriks et al. Nat Biotech, 2023, doi: 10.1038/s41587-023-01680-4.
6. Rüland, ...., Hendriks#, Artegiani# Nat Commun, 2023, doi: 10.1038/s41467-023-37951-6.
7. Honeyman et al. Science 343(6174) 1010-1014, 2014.
8. Hirsch et al. J Hepatol 72(5) 924-936, 2020.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in