Mapping electron transfer trajectory to multiple vibrational reaction coordinates
Published in Chemistry
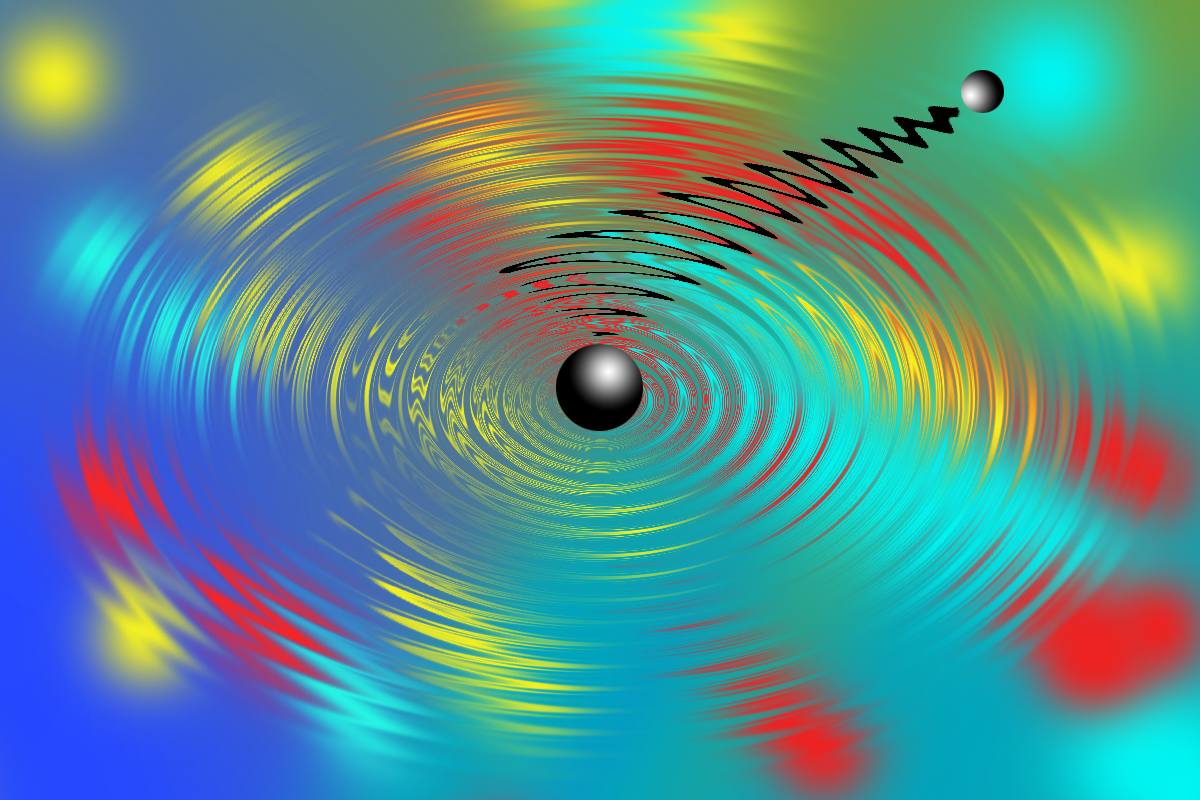
Molecular vibrations represent an ordered or synchronized beating of the molecular structure at well-defined periodicities. These periodic beatings of the molecular structure have been increasingly recognized to couple with electronic dynamics and control light-induced function of molecular systems on ultrafast timescales. For example, they are responsible for: enabling superfast isomerization of retinal in protein rhodopsin, which is the primary step in the process of vision; photodissociation of diatomic systems; ring opening in cyclohexadiene; and nonradiative curve-crossing funnels; etc.1 These remarkable studies cater to this revolutionary idea of controlling reactivity and dynamics by exploiting the quantum mechanical order provided by synchronizing molecular vibrations and electronic trajectory.
In our Nature Chemistry work,2 we resolve a cascade of quantum mechanical events in the reaction trajectory for a ballistic electron transfer (ET) reaction, providing a fingerprint of how molecular structure warps sequentially along a reaction path in response to a reactive event. The ultimate revelation is that there is an order to the structural changes associated with a reaction that is decided by the frequencies of the vibrational modes.
Electron transfer reactions are vital to vastly diverse processes ranging from photosynthesis to redox reactions and solar energy technology to molecular electronic devices, therefore modern chemistry pursues ways to understand and control these reactions. Electron-vibrational interplay can provide that quantum mechanical pivot.3,4 An experimental approach to exploring the intricate electron-vibrational dynamics in functional systems, that has received significant traction over the last two decades, requires that all the light-absorbing entities are synchronized in-step. This synchronization is made possible by using resonant laser pulses in the visible spectrum compressed to ~10-femtoseconds in state-of-the-art laser spectroscopic techniques.5 Besides populating the high-energy electronic states, these laser pulses also create coherent superpositions of vibrational levels along vibrational modes whose equilibrium position is displaced between the low- and high-energy electronic states. These superpositions of vibrational levels evolve as wavepackets/coherences on electronic potentials and relay the information back to us by creating time-dependent periodic amplitude modulations of the nonlinear signal. The period of those modulations corresponds to the frequency of the vibrations. The simultaneous probing of electronic population and vibrational wavepackets provides a platform that allows the elucidation of electron-vibrational phenomenon.
A grand challenge for researchers in this community, however, is how to identify vibrational coherences relevant to the ET reaction from the vast number of coherences generated along various Frack-Condon active vibrations, the majority of which are spectators.6-8 In addition, can vibrational coherences reveal anything about the reaction coordinate? Early on, we approached these challenges by researching the role of vibrations in various ET systems; for example, forward ET reaction between an organic acceptor Oxazine 1 and donor dimethylaniline, intramolecular backward ET in Betaine-30 in methanol and acetonitrile.9,10 The study of these systems helped us formulate ground rules – both mechanistic and technical – for a potential system that could help us distinctly identify the interplay of electronic and vibrational degrees of freedom. Our conditions were, 1) that the ET reaction should be superfast (significantly faster than the dephasing of the vibrational coherences), 2) that the ET should be in the Marcus-inverted region, and 3) that the ET product must absorb in the visible spectral region. We were also able to postulate a hypothesis that ET reaction induces dephasing of the vibrational coherences along the modes coupled to the ET reaction.7
The above conditions were addressed by using perylenetetracarboxylic diimide (PDI) as electron acceptor and dimethylaniline as electron donor. In this model ET system, forward ET reaction occurs in ~30-femtoseconds and is in the Marcus inverted region, and the PDI radical anion absorption band lies in the visible spectrum. We discovered, in our data, the abrupt loss of phase coherence along some high-frequency vibrational coordinates associated with the product state. This rapid loss of phase coherence along the high-frequency vibrations originates from the random phase interference of ET reaction pathways and directly reports on the vibrationally driven reaction trajectory from the reactant state to the transition state. This observation is contrary to conventional Marcus theory, where stochastic fluctuations of the solvent promote ET by creating energetic disorder, whereas, here, it is the high-frequency vibrations driving the ET reaction. This experimental observation was not only able to explain the superfast ~30-femtosecond time constant of the reaction, but also confirmed our above hypothesis that ET reaction induces dephasing of the vibrations coupled to the reaction.
When we started working on this problem two years ago, an experimental observation that surprised us the most and kept us on our toes for a few months was a completely unexpected low-frequency wavepacket. To put it in context, we use laser pulses to create wavepackets in molecules, whereas this “unexpected wavepacket” did not seem to be generated by the laser pulse. This astonishing observation was followed by a series of control experiments, which enabled us to speculate and hypothesize, that this unexpected wavepacket may in fact be created by the superfast ET reaction itself, and not by the laser pulse. That is when we turned to quantum dynamics simulations. These simulations helped us establish that, as soon as the system reaches the transition state owing to the high-frequency vibrations, the acceptor molecule responds by instantly shifting the equilibrium position of a low-frequency vibration. This sudden shift in equilibrium position along a vibrational mode in response to the superfast ET is akin to stretching a vibrating spring to a more stable position, with an added property that the spring is now vibrating with a significantly larger amplitude. This spring-like response of the synchronized beating of the molecular structure to the ET provides a sink that inhibits coherent recurrence of the ET that might otherwise be expected for a ballistic process.
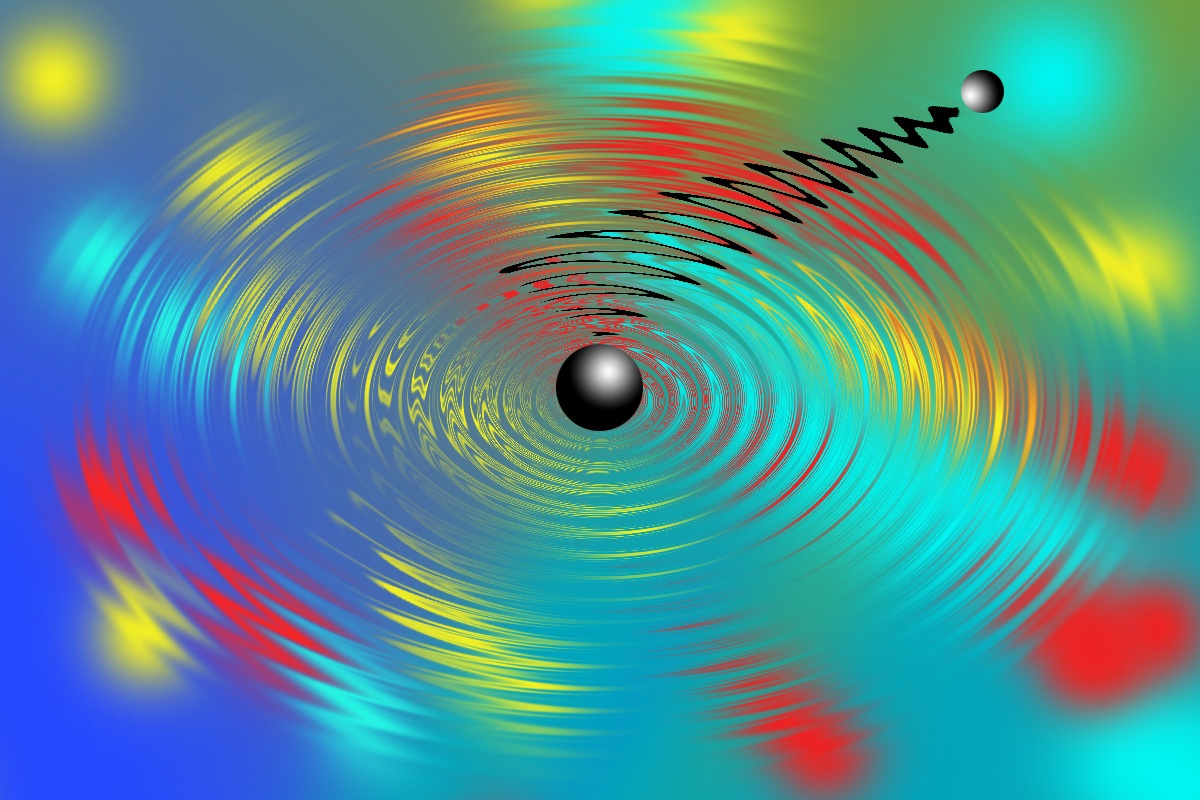
In summary, we discovered that high- and low-frequency beatings of the molecular structure interplay, so the ET trajectory maps to multiple vibrational reaction coordinates. This allows for a sequence of structural reorganization and relaxation stages accompanying the ultrafast change in charge distribution post ballistic ET. Thereby, the challenge of identifying reaction-relevant vibrational degrees of freedom was overcome and the role of vibrational coherences in unravelling the path of the reaction trajectory and composition of the reaction coordinates was established.
References:
- Hoffman, D. P. & Mathies, R. A. Femtosecond Stimulated Raman Exposes the Role of Vibrational Coherence in Condensed-Phase Photoreactivity. Acc. Chem. Res. 49, 616-625 (2016).
- Rafiq, S., Fu, B., Kudisch, B. & Scholes, G. D. Interplay of vibrational wavepackets during an ultrafast electron transfer reaction. Nat. Chem. (2020). https://doi.org/10.1038/s41557-020-00607-9
- Delor, M. et al. Toward control of electron transfer in donor-acceptor molecules by bond-specific infrared excitation. Science 346, 1492-1495 (2014).
- Bredas, J. L., Sargent, E. H. & Scholes, G. D. Photovoltaic concepts inspired by coherence effects in photosynthetic systems. Nat. Mater. 16, 35-44 (2017).
- Rafiq, S. & Scholes, G. D. Slow Intramolecular Vibrational Relaxation Leads to Long-Lived Excited-State Wavepackets. J. Phys. Chem. A 120, 6792-6799 (2016).
- Rafiq, S., Bezdek, M. J., Chirik, P. J. & Scholes, G. D. Dinitrogen Coupling to a Terpyridine-Molybdenum Chromophore Is Switched on by Fermi Resonance. Chem 5, 402-416 (2019).
- Rafiq, S. & Scholes, G. D. From Fundamental Theories to Quantum Coherences in Electron Transfer. J. Am. Chem. Soc. 141, 708-722 (2019).
- Scholes, G. D. et al. Utilizing Coherence to Enhance Function in Chemical and Biophysical Systems. Nature (London) 543, 647-656 (2017).
- Rafiq, S., Dean, J. C. & Scholes, G. D. Observing Vibrational Wavepackets during an Ultrafast Electron Transfer Reaction. J. Phys. Chem. A 119, 11837-11846 (2015).
- Rafiq, S. & Scholes, G. D. Is back-electron transfer process in Betaine-30 coherent? Chem. Phys. Lett. 683, 500-506 (2017).
Physical Chemistry, Ultrafast Spectroscopy, Solar-energy conversion, Optoelectronic molecules and materials, Electron transfer, Quantum information, Quantum coherences, Excited state dynamics, Triplet harvesting, Two-dimensional materials, Coherence spectroscopies, Two-dimensional electronic spectroscopy
Follow the Topic
-
Nature Chemistry

A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in