Poster Presentation: Bringing interaction-state proteomics to the web with the dfPPI Analysis Platform at Next Gen Conversations, 2026
Published in Cancer, Neuroscience, and Protocols & Methods

Looking beyond protein abundance
A central idea behind our poster is simple: knowing how much of each protein is present does not always tell us how the cell is functioning.
Cellular processes are organized through protein–protein interactions, or PPIs. These interactions form the dynamic functional wiring of a cell. Under disease-associated stress, proteins may lose their appropriate interacting partners, gain abnormal interactions or become incorporated into pathogenic assemblies. The resulting changes can rewire entire pathways, sometimes without corresponding changes in the abundance of the proteins themselves.
Most transcriptomic and conventional proteomic methods are designed primarily to measure abundance. The dysfunctional Protein–Protein Interactome, or dfPPI, framework addresses a complementary question:
How does a protein’s engagement with cellular interaction networks change between biological conditions?
By focusing on interaction state rather than abundance alone, dfPPI provides a functional view of how cellular networks become disrupted in disease.
From interaction capture to systems-level biology
dfPPI combines interaction-capture chemoproteomics, mass spectrometry and network-based computational analysis.
Selective chemical probes are used to capture disease-associated protein assemblies from native biological samples. Mass spectrometry then identifies and quantifies the proteins associated with these assemblies. The computational workflow compares the resulting measurements across conditions, and models changes in protein interaction engagement.
Rather than attempting to produce a static catalogue of every binary protein interaction, dfPPI measures how effectively proteins are recruited into interaction networks at the systems level. These changes can then be interpreted through biological pathways, functional modules and disease-associated network states.
The approach can be applied to disease models and native tissues, including patient-derived samples, providing a way to investigate network-level dysfunction in biologically relevant contexts.
Why build a web-based analysis platform?
As the dfPPI framework developed, its computational analysis was standardized and refined across multiple studies. However, a complete analysis still requires several coordinated steps, including:
- dfPPI-specific data transformation and normalization;
- handling of missing mass-spectrometry measurements;
- differential interaction-engagement modelling;
- pathway enrichment and network interpretation;
- visualization and export of results.
Together, these steps can represent a substantial barrier for researchers who do not routinely work with computational pipelines.
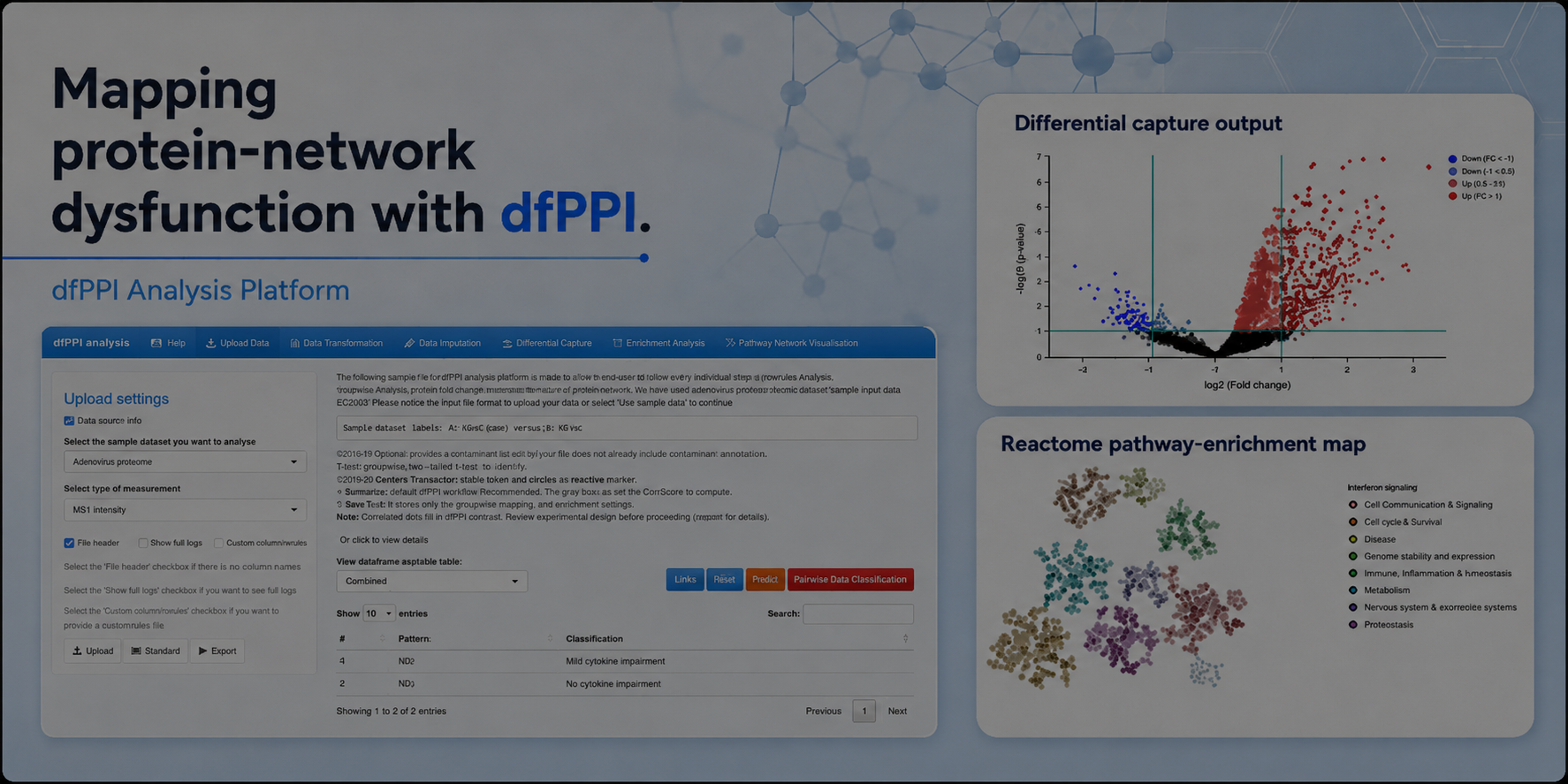
The dfPPI Analysis Platform brings the validated workflow together in a point-and-click web interface. Users upload a dfPPI interaction-capture mass-spectrometry intensity table and select one of two analysis modes.
In Standard mode, the platform applies literature-validated default settings and guides users through the complete analysis. This mode is intended to make the workflow accessible to researchers who may have limited programming or bioinformatics experience.
In Expert mode, users have greater control over the analytical parameters. Detailed modelling steps, processing logs, interactive visualizations and downloadable intermediate results are available to support specialized computational workflows and closer inspection of the analysis.
Built-in example datasets and stepwise guidance also allow users to explore the platform before uploading their own data.
The resulting outputs include differential-capture results, pathway-enrichment maps, pathway-rewiring visualizations, interaction-state vulnerability profiles and downloadable result tables. The aim is not only to reduce analysis time, but also to promote transparent and reproducible interpretation of dfPPI experiments.
What has dfPPI revealed so far?
The poster places the new web server in the context of previous biological applications of dfPPI.
In cancer, systems-level dfPPI analyses have identified protein-network dysfunctions that allow tumour cells to adapt to stress and maintain malignant phenotypes. These analyses revealed that different cancer states can depend on distinct configurations of cellular interaction networks.
In Alzheimer’s disease, dfPPI uncovered interaction-state dysfunction associated with hippocampal stress and impaired synaptic plasticity. These findings demonstrated that neurological dysfunction can be associated with changes in protein connectivity that are not readily apparent from protein-expression measurements alone.
Pharmacological studies have also shown how manipulating disease-associated interaction networks can produce therapeutic vulnerability. At the same time, treatment can trigger network reorganization and cellular rebound through the reactivation of pre-existing protein pathways, providing a systems-level explanation for both treatment response and resistance.
Together, these studies illustrate an important principle: disease phenotypes can be encoded in protein connectivity, not only in protein abundance.
The main take-home message
One of the most striking lessons from dfPPI studies is that phenotype-specific dysfunction can arise through stressor-specific remodelling of the protein interaction state.
A protein does not necessarily need to disappear or become strongly overexpressed to acquire an important role in disease. A change in its interaction partners—or in the network into which it is recruited—may be sufficient to alter cellular function.
Abundance-based omics therefore remains essential, but it represents only one layer of disease biology. Interaction-state proteomics adds information about how the molecular components of a cell are functionally organized.
By making the analysis more accessible and standardized, the dfPPI Analysis Platform is intended to help researchers include this additional layer in routine disease investigations.
Reflections from NextGen Conversations
The Next Gen Conversation in Biomolecular Structure and Dynamics was a particularly fitting setting in which to present this work.
Research on biomolecular structure and dynamics often examines how individual molecules change their conformations, assemblies and interactions. dfPPI connects these molecular events to a larger scale: chemical biology captures native protein assemblies, mass spectrometry measures their components, and computational analysis translates those measurements into pathway- and network-level models.
The platform also addresses a recurring challenge in computational biology: how can a sophisticated analysis become easier to use without turning into a black box?
Our two-mode design is one response to that challenge. Standard mode provides an accessible and reproducible route through the analysis, while Expert mode preserves detailed control and transparency for computational users.
Presenting the platform alongside experimental, structural and computational studies highlighted the importance of connecting these different scales of biological organization—from molecular assemblies to cellular pathways and disease phenotypes.
What comes next?
The immediate goal is to encourage broader use and evaluation of the platform across different disease models, biological samples and experimental designs.
Community feedback will be important for refining the interface, documentation, visualizations and analytical options. Wider application will also help establish where interaction-state measurements provide the greatest additional value over conventional expression-based analyses.
In the longer term, integrating dfPPI with transcriptomics, conventional proteomics and other molecular measurements could provide a more complete picture of how a biological stressor becomes cellular dysfunction—and where that dysfunction may create a therapeutically actionable vulnerability.
The dfPPI Analysis Platform is available at https://dfppi.mskcc.org/. We welcome researchers to explore the example datasets, test the workflow and share their feedback.
Acknowledgements
This dfPPI Analysis Platform was developed by Souparna Chakrabarty, Tai Wang, Vlad-Cristian Miclea, Radu Ioan Peter, Shambhavi Joshi, Manda Wilson, Nicholas Socci and Gabriela Chiosis.
The project brings together expertise in chemical biology, mass spectrometry, bioinformatics, mathematics, computer science and disease biology across Memorial Sloan Kettering Cancer Center, the Technical University of Cluj-Napoca and New York University.
Selected references
- Chakrabarty, S., Wang, S., Roychowdhury, T., Ginsberg, S. D. & Chiosis, G. (2024). Introducing dysfunctional Protein–Protein Interactome (dfPPI)—A platform for systems-level protein–protein interaction dysfunction investigation in disease. Current Opinion in Structural Biology, 88, 102886. doi: 10.1016/j.sbi.2024.102886.
- Rodina, A. et al. (2025). Mapping Dysfunctional Protein–Protein Interactions in Disease. Journal of Visualized Experiments, 224, e69197. doi: 10.3791/69197.
- Rodina, A. et al. (2023). Systems-level analyses of protein–protein interaction network dysfunctions via epichaperomics identify cancer-specific mechanisms of stress adaptation. Nature Communications, 14, 3742. doi: 10.1038/s41467-023-39241-7.
- Joshi, S. et al. (2021). Pharmacologically controlling protein–protein interactions through epichaperomes for therapeutic vulnerability in cancer. Communications Biology, 4, 1333. doi: 10.1038/s42003-021-02842-3.
- Inda, M. C. et al. (2020). The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivity-based dysfunction. Nature Communications, 11, 319. doi: 10.1038/s41467-019-14082-5.
- Rodina, A. et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature, 538, 397. doi: 10.1038/nature19807.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in