Return of the chain gang: infinite protein filaments in evolution and disease
Published in Microbiology

‘Molecular medicine’, an approach that understands human disease as caused by malfunctioning molecules that make up the human body, dates back to the 1949 paper by Linus Pauling Sickle Cell Anemia, a Molecular Disease.
By using electrophoresis, Pauling, Itano, Singer, and Wells showed that haemoglobin from healthy individuals and from patients suffering from (a particular subtype of) sickle-cell anaemia differed in electrical charge. Electrophoresis – its early ‘moving boundary’ variety – was then a relatively novel method, just awarded a Noble prize a year earlier, and it took precision to perform it well, but it was nonetheless simple: you could make an electrophoresis apparatus in your garage. And it was the relative simplicity of the study that reinforced its paradigm-changing message. A reader would put the article down with the impression that the complicated reality of human disease might in the end boil down to basic physicochemical properties of specific, individual proteins. The impression that inspired, for good and for bad, medical and life sciences ever since.
8 years later, and exactly 60 years ago this month, Vernon Ingram, one of the pioneers of protein sequencing, reported that the difference between the normal and the disease-causing haemoglobin forms was exactly one amino acid; in the protein isolated from sickle-cell anaemia patients, one particular glutamate residue in subunit beta was replaced with a valine. As haemoglobin is composed of two alpha and two beta subunits, there were two mutations per a functional molecule. This was sufficient to trigger aggregation of haemoglobin into long, insoluble, crystal-like filaments that interfered with normal erythrocyte function, causing disease. In Ingram's words,
[t]he abnormally low solubility of reduced haemoglobin S, which causes the sickling of the erythrocytes in the anaemia, is presumably a function of the charge distribution on the surface of the molecule. The replacement of two charged glutamic acid residues for two uncharged valines is presumably enough to alter this distribution towards one favouring abnormally easy crystallization.
Ingram’s hypothesis – that such small, single-amino-acid changes on protein surfaces might ‘favour abnormally easy crystallization’ – echoes in a study published two weeks ago by the group of Emmanuel D. Levy, a study performed with E. coli protein complexes and thus, I hope, qualifying for this Nature Microbiology Communities blog.
If a mutation on a protein surface creates a sticky, self-interacting patch, then this is expected to produce one of two possible outcomes. If a protein is monomeric, two copies of it would stick to each other, burying their self-interacting patches and creating a finite dimer. If, however, a protein is a symmetrical complex of two or more proteins encoded by the same gene – as in the case of haemoglobin – this would result in a formation of an infinite – or at least very long – filament.
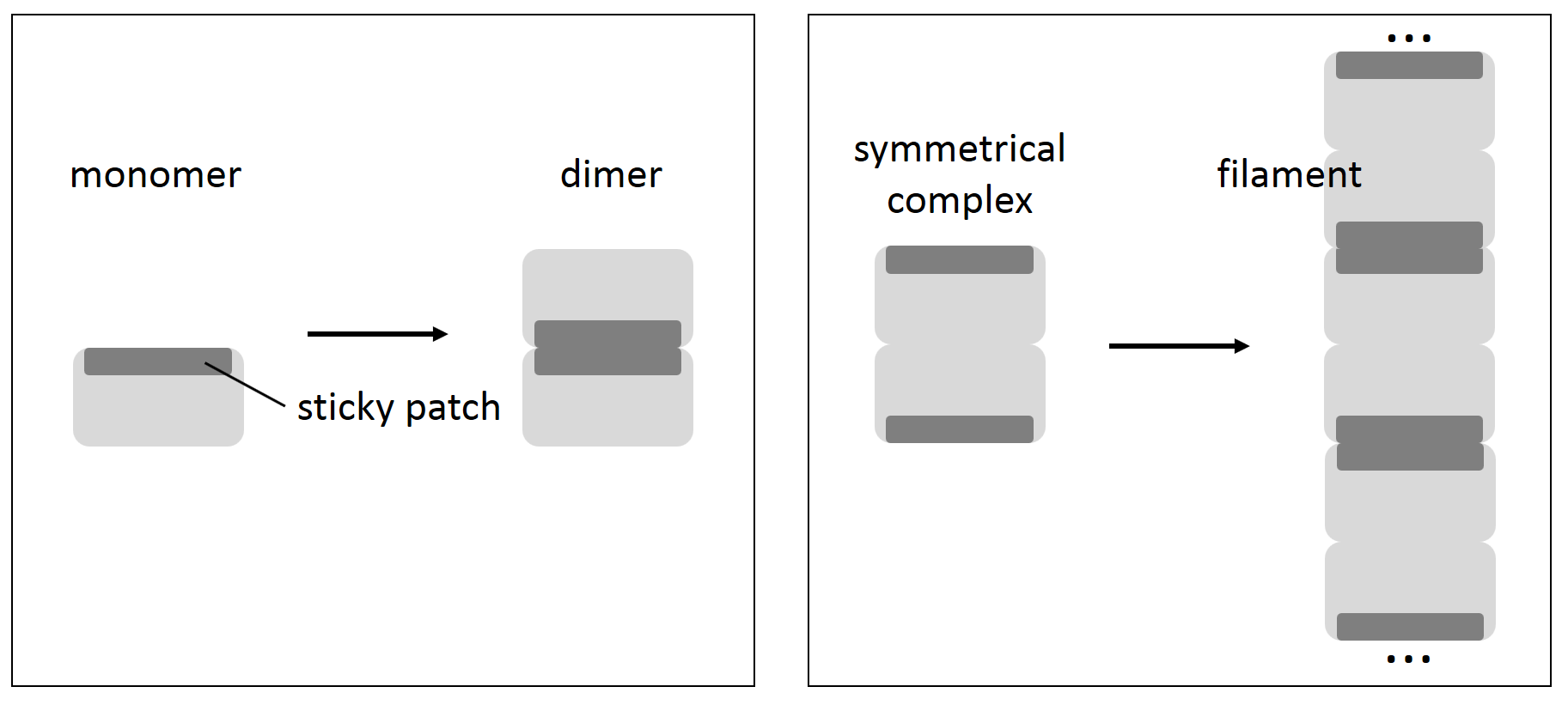
What Garcia-Seisdedos, Empereur-Mot, Elad, and Levy now show is just how easy creating such sticky patches is. Mutating small numbers of charged surface residues to either leucines or tyrosines in each of 12 different E. coli symmetrical complexes taken for the analysis resulted in the formation of long chains of folded molecules. In 4 cases, just one mutation was sufficient to turn a small complex into an almost infinite supramolecular assembly.
This, of course, has evolutionary implications. It seems that symmetrical complexes, while having many advantages (e.g. allostery, frugal genetic encoding of large structures), are always on the edge of turning into long insoluble chains. Avoiding this risk might be one of the reasons behind the transition from symmetrical homocomplexes to quasi-symmetrical heterocomplexes observed in higher organisms (think the 20S proteasome in actinobacteria vs. in yeast or humans). Wherever symmetrical complexes are present, especially in fast-evolving lower organisms, filamentous states are expected to be frequently sampled by evolution, and – although, as Levy and coworkers show, usually are selected against – might sometimes confer functional advantage.
Finally, authors are right that aggregation and insolubility – often spotted by various methods in vivo and in vitro – should not be ‘dogmatically interpreted in the context of misfolding’. And not necessarily in the context of disease, although the sickle-cell anaemia-causing mutant haemoglobin remains the most famous example.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in