Sleep as an Early Signal in ASD Genetics: Insights from Juvenile Shank3-Deficient Rats
Published in Neuroscience, General & Internal Medicine, and Behavioural Sciences & Psychology
Why we focused on sleep in the first place
In autism spectrum disorder (ASD), sleep problems are not a small “side issue.” They are often among the earliest difficulties parents notice—and among the hardest to manage. Clinical studies suggest that roughly 50–80% of children with ASD experience some degree of sleep disturbance; in children carrying SHANK3-related variants, caregiver-reported sleep problems can be even more common, reaching ~90% in some cohorts. Crucially, these difficulties often emerge early in life and intertwine with daytime emotion regulation, attention, and behavior, sometimes forming a cycle that amplifies challenges on both sides.
This raises a central scientific question: are sleep problems merely a downstream consequence of ASD, or are they an early, intrinsic phenotype of certain genetic subtypes? If the latter is closer to the truth, then sleep becomes more than a symptom. It becomes an early, measurable, and potentially actionable feature—one that could open a realistic path toward early intervention.
That framing also implies something practical that the field still lacks: a tractable juvenile-stage animal model with objective physiological readouts, where we can study early ASD-relevant mechanisms and test interventions during a developmental window that matches the clinic. Working on Shank3, a high-confidence monogenic ASD risk gene, we kept returning to the same hypothesis: can genetic risk shape sleep regulation itself early in development? If so, sleep would no longer be only the “shadow” of behavioral phenotypes, but a mechanistically informative signal we can track longitudinally—and potentially modify.
Why juvenile sleep phenotyping matters
Many preclinical Shank3 sleep studies focus on adulthood, whereas clinically, sleep problems often appear much earlier. This mismatch in timing motivated us to shift our window to the juvenile stage, using Shank3-deficient rats to address three questions that better reflect clinical reality:
-
Does sleep disruption already emerge during development?
-
Beyond “how long they sleep,” does it alter sleep architecture and sleep depth ?
-
When sleep is deprived, can the system still mount a normal homeostatic rebound?
Juvenile EEG/EMG recordings are not easy: the animals are smaller, signals are more sensitive, surgeries and recovery require greater precision, and long-term stability is harder to maintain. Over time, we realized that a major part of this project was not a single “analysis trick,” but building a dataset we could trust—stable recordings, reliable scoring, and consistent readouts—before we tried to interpret mechanisms.
From a translational perspective, juvenile phenotyping is also more than simply “looking earlier.” If sleep abnormalities are already present during development, they align with a real-world early-intervention window: many families seek clinical help for sleep long before core ASD symptoms fully crystallize. Sleep is one of the few targets that can genuinely align the “research window” with the “clinical window.”
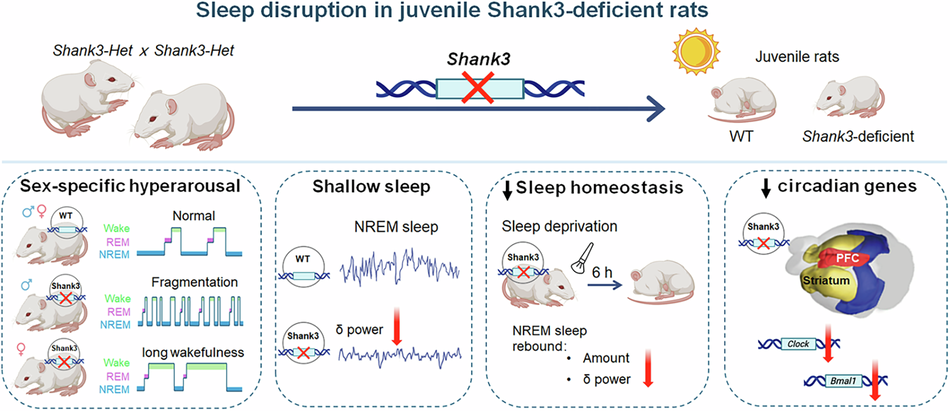
The finding that surprised us most: one theme, two sex-divergent patterns
We initially expected a relatively uniform sleep deficit—for example, an overall reduction in total sleep. Instead, what stood out most was sex divergence. Both male and female Shank3-deficient rats showed a hyperarousal-like sleep profile, but the manifestations separated into two distinct patterns:
-
Male mutants: more pronounced sleep fragmentation, suggesting impaired sleep maintenance—closer to “light, easily broken sleep.”
-
Female mutants: prolonged wakefulness, resembling difficulty falling asleep again after arousal, or problems with sleep initiation/re-initiation—closer to “once awake, hard to return to sleep.”
This matters because it suggests that the same genetic risk factor may map onto different “insomnia-like” subtypes. Clinically, that feels familiar. Scientifically, it pushed us to treat sleep disruption as a structured phenotype, not something to be summarized by a single endpoint such as total sleep time. It also implies that future interventions may need to be more precise, matching strategies to the dominant problem (maintenance vs initiation/re-initiation) rather than applying a one-size-fits-all approach.
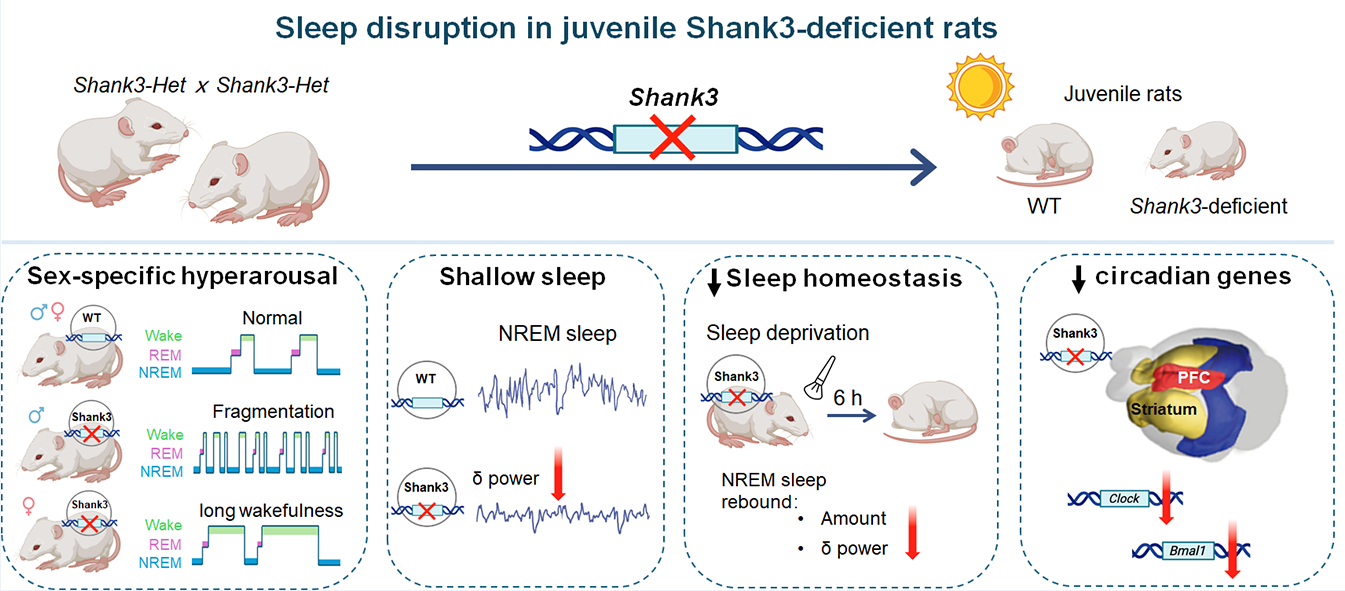
Shallow sleep—and weaker recovery—may matter more than “a bit less sleep”
Sleep architecture tells us how sleep is organized, but EEG power spectra speak more directly to how deep sleep is. During NREM sleep, we observed reduced delta (δ) power, commonly linked to slow-wave activity and restorative sleep. This suggests the phenotype is not merely a change in sleep duration, but also a change in sleep depth—a quality dimension that can be underestimated if one focuses only on time asleep.
We then challenged sleep homeostasis with 6 hours of sleep deprivation. Under normal conditions, sleep loss triggers compensatory increases in NREM sleep and δ power during recovery—an adaptive rebound. In juvenile Shank3-deficient rats, this rebound was blunted: they appeared less able to “pay back” sleep debt.
For us, this result was particularly important for early intervention. If a developing brain experiences chronically shallow sleep and has a weakened capacity for homeostatic recovery, the consequence may not be one or two bad nights, but a persistent shift in the developmental milieu that accumulates over weeks and months. This is exactly where early intervention becomes meaningful: the goal is not simply “more sleep,” but improving sleep depth and homeostatic recovery dynamics during a period when neural circuits remain highly plastic—potentially reducing downstream cascading effects at their source.
A molecular clue: circadian gene downregulation in the corticostriatal system
To connect these phenotypes to a trackable biological axis, we examined circadian-related gene expression. We found downregulation of Clock and Bmal1 in regions including the prefrontal cortex and striatum. This does not, by itself, complete a mechanistic pathway. However, it provides a coherent molecular direction consistent with the behavioral and electrophysiological phenotype and suggests circadian dysregulation within corticostriatal systems—offering a clearer route for the next phase of mechanistic work.
What we hope readers take away
The core value of this work is both developmental and translational. By demonstrating early-onset, quantifiable sleep disruption in juvenile Shank3-deficient rats—spanning sleep architecture, EEG-defined sleep depth, and homeostatic regulation—we establish a tractable preclinical model that aligns with the timing of sleep problems in children with ASD. In other words, this is not only a set of observations in a known ASD gene model: it provides a platform to study juvenile-stage ASD mechanisms in a window when neural circuits are still maturing, and to test early, sleep-focused interventions with objective physiological readouts.
More broadly, our framework shifts attention from “sleep amount” to architecture + depth + homeostasis, dimensions that may better capture clinically meaningful sleep dysfunction. The sex-divergent patterns we observed further suggest that a single genetic risk factor can manifest as distinct insomnia-like subtypes, motivating mechanism-driven, subgroup-aware intervention strategies.
Looking ahead, we aim to map the circuit nodes that drive hyperarousal and impaired homeostasis, unpack the biological basis of the sex divergence, and determine whether intervening during this juvenile window can reshape downstream behavioral and cognitive trajectories. In developmental neuroscience, sleep should not be treated as background noise—especially for genetic subtypes where it may be an early signal and a practical entry point for change.
Follow the Topic
-
Translational Psychiatry
This journal focuses on papers that directly study psychiatric disorders and bring new discovery into clinical practice.

Related Collections
With Collections, you can get published faster and increase your visibility.
Moving towards mechanism, causality and novel therapeutic interventions in translational psychiatry: focus on the microbiome-gut-brain axis
Publishing Model: Open Access
Deadline: Nov 15, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in