
Genesis
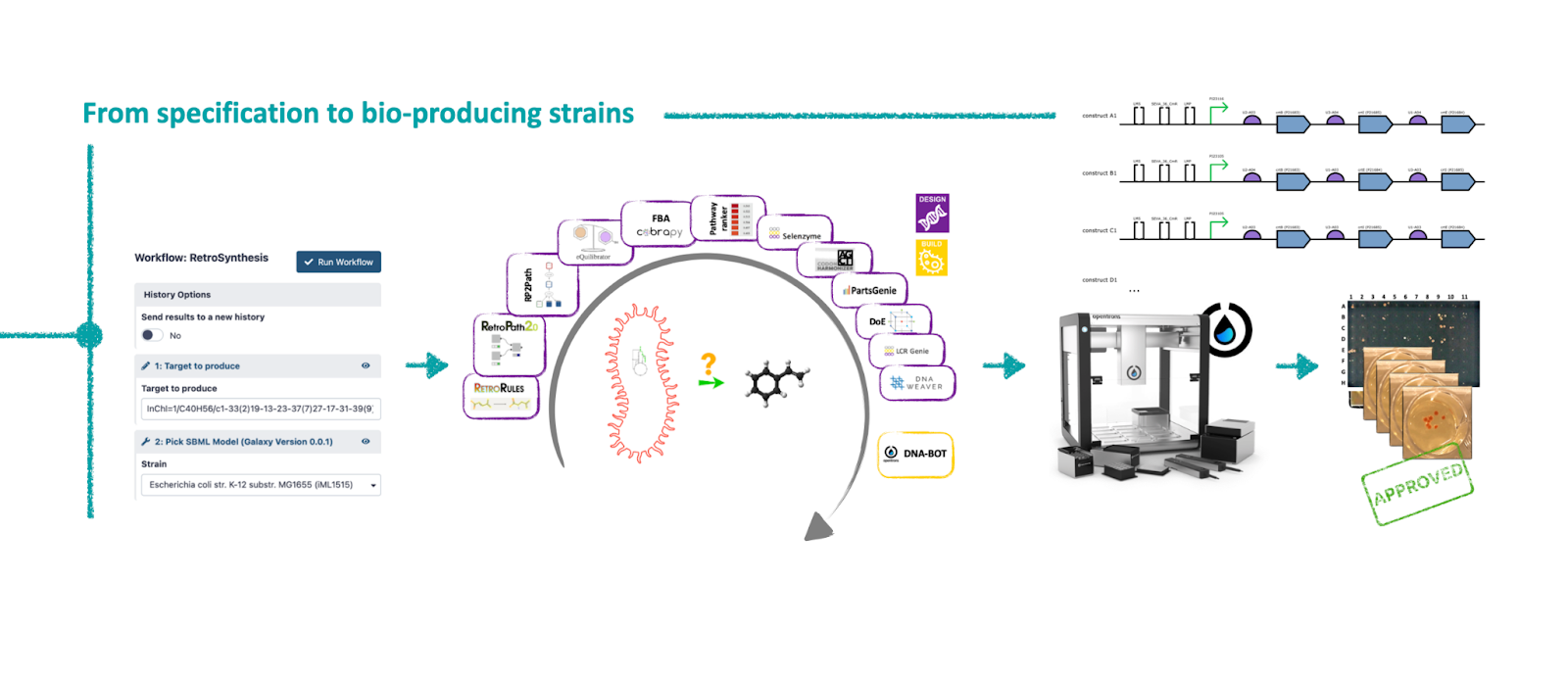
In 2019, the development process of a Synthetic Biology project was already well defined by the Design-Build-Test-Learn paradigm. There were many tools available to answer questions like: "Can I engineer this microorganism to produce that compound?". Our research group at INRAE had developed some of them (RetroRules, RetroPath2, RP2Paths...). Once installed, the tools worked well together because the file formats were designed so that they could communicate with one another. But as soon as we used a tool developed by someone else, we ran into several challenges. The first one was the installation problem because we needed the right versions of libraries and sometimes the right versions of OS. Once installed, the second challenge was communication between the tools, which did not all use standard formats. A third challenge emerged as we used more tools: orchestrating the execution of multiple tools and managing exchange files. Indeed, as our goal was to be able to modify an organism to produce a molecule of interest, we were using about fifteen different tools developed by half a dozen different research groups.
At the same time, the Galaxy system was mature and the community already well established. This system offered the possibility of chaining tools together through a simple graphical interface and pressing a button to start the workflow. In fact, any user could run a series of tools from their web browser without installing them or worrying about managing the exchange files. Galaxy would allow us to integrate all the tools we used in a single interface in order to democratize their use.
All we had to do was to et to work. While Galaxy-SynbioCAD is easy to use, it was not easy to develop. Here is the take of the programming geeks that made this happen.
Entering into the Galaxy
The initial plan was to encapsulate programs in Docker images and run them with Galaxy. The idea was appealing, as Docker makes installations easier, helps with runtime reproducibility, and offers great implementation flexibility for encapsulated tools.
Unfortunately, we realized that using Docker was not suitable for all Galaxy instances, especially on high performance computing infrastructures where the recommended route was the conda package manager. This became apparent in discussions with administrators of national Galaxy platforms (such as https://usegalaxy.fr/), for whom the implementation of Docker-based wrappers posed security and implementation concerns. While we had already encapsulated several tools with Docker, we revised our strategy and abandoned Docker technology in favor of the conda packaging system to maximize the possible deployment sites for the tools.
At the same time, we were writing tests for the programs we were developing in our research group and setting up continuous development and integration mechanisms. This allowed us to publish the packages on community channels (conda-forge and bioconda). Once a program was available as a conda package, a wrapper had to be written to integrate it into the Galaxy system. Here again, tests were conducted for each wrapper written to ensure that it would work properly in Galaxy before being integrated into the Galaxy ToolShed – a sort of App Store for Galaxy that allows tools to be installed from any Galaxy instance.
We did this work for all the tools that our research group had developed but also for all the tools developed by our partners involved in this crazy project. For some tools, we had to make the program executable without the need of a graphical interface (because the Galaxy wrapper uses command line instructions), while continuing to offer the same options, the same functionalities, and while continuing to offer the graphical version as an alternative. All this work took a substantial amount of time requiring one person working full time on the conda packaging, publication on conda-forge or bioconda, as well as on writing and testing the wrappers and finally publishing these on the Galaxy ToolShed.
Exploring the Galaxy
Once all the tools were available on the Galaxy system and the different workflows were created, we conducted a multi-site study to demonstrate that the system could really run from anywhere and that it was simple to do. This collaboration, mixing (i) code writing (with its share of commits, pull requests, bugs), (ii) protocol development (with its share of technical questions, product references, ...) and (iii) exchanges of biological material (plasmid shipments) was very exciting as Mahnaz, leader in Micalis for the bench part, testifies:
“In this study, I contributed to benchmarking workflows for Lycopene production. While working on lycopene pathway gene assembly, it was the first time I met our liquid handler robot Opentrons that we named OTTO! And indeed, it was a delightful encounter. Thanks to OTTO — after using Galaxy-SynBioCAD Genetic Design (BASIC) workflow that “translated” the best predicted pathway into Opentrons instructions — we did automated construction of 88 distinct plasmids following the BASIC assembly method. I had an excellent experience collaborating with Gizem and Geoff from Imperial College London. We performed the biological replicates in two laboratories in London and Paris in parallel. In the last step performed by OTTO we encountered some challenges and thanks to this collaboration, the optimization process went flawlessly!”
During this investigation for lycopene production, the Galaxy SynBioCAD was really helpful compared to what we would have to do if it was not there, such as searching the pathways and enzymes in literature, and writting manual assembly plans. The “on the cloud” nature of Galaxy also makes much easier exchanging results between collaborators.

The journey for storing annotations
During the project, we had several discussions about how to store the information we needed for metabolic pathway reconstruction and classification. This includes, for example, the chemical structures of the compounds in InChI form, or the identifiers of the rules needed for pathway completion. There were also considerations of where to store the results of pathway analyses, such as FBA fluxes, pathway Gibbs free energies, and global scores. We once considered associating each SBML file modeling a pathway with a JSON annotation file that would contain the information not natively present in the SBML. However, this would have made it more challenging to manage files between tools. As soon as we considered the analysis not of a pathway, but of a collection of pathways, it was also necessary to manage the matching between SBML and JSON files. Finally, we came to the conclusion that managing everything in an SBML file using annotation fields, in the way MIRIAM annotations are done, would be more portable and more convenient to communicate with other programs, in a word: more FAIR. This is what we did, with what we called between us (and then in the paper) the "rpSBML" which correspond to files in SBML format with annotation sections (for chemical species and reactions) and annotation sections enriched in information. A strength of rpSBML is that as the SBML specifications are respected, they are compatible with any tool able to manage SBML files. We had also considered extending the SBML specifications with the development of a new package (as is the case with the SBML package "fbc" to manage FBA information), but the amount of additional work was significant and required an advanced knowledge of SBML package creation. We preferred not to take the step (for the moment).
At the end it was a substantial coding effort but we believe we created a tool that is easy to use for all, especially those working at the bench not necessarily familiar with all the conda, Docker, Python jargon, and we recommend to follow our tutorials published on the Galaxy training web page: https://training.galaxyproject.org/training-material/topics/synthetic-biology/.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in