Turning gas pipeline toward chemical synthesis: catalyst-free conversion of methane to methanol and hydrogen by water radical cations
Published in Chemistry

Just like in the days of our extinct ancestors, Homo erectus, methane (CH4), the major component of natural gas, is still predominantly utilized by the 21st century man simply as fuel for heat generation. However, the global ecological implications of CH4 burning are increasingly alarming and cannot be further neglected: since the industrial revolution, this process accounts for approximately 30% of the global temperature rise due to the release of carbon dioxide (CO2). In the coming era of carbon-free energetics, a grand challenge for mankind is to master the utilization of CH4 for chemical synthesis, thus redirecting it from mere combustion towards the conversion into valuable chemicals and materials. Similar redirection toward chemical synthesis of polymers has been largely accomplished for natural oil, and it is now the turn for natural gas and CH4.
However, the efficient conversion of CH4 to oxygenated chemicals remains a formidable task for industry. Due to the very high stability and inertness of CH4 molecule, harsh conditions such as high temperatures and pressures are normally required to activate its C–H bonds. This greatly increases capital investment and leads to operational risks and environmental problems. Another key problem that blocks the road toward efficient CH4 chemistry is the overoxidation of CH4 oxidation products (e.g., methanol, CH3OH), which is extremely difficult to control, particularly in catalytic systems, and which greatly compromises the yield and selectivity of CH4 conversion and leads to the poorly controllable and undesirable combustion to CO2. Obviously, principally new approaches are needed for the cost-efficient conversion of CH4.
Over recent years, using ambient corona discharge ionization of water vapor, in our group we learned to generate highly reactive water dimer radical cations, (H2O)2+•. The possibility to accelerate these ions by non-sophisticated equipment makes them ideal candidates for unorthodox chemical transformations otherwise unachievable at room-temperatures. The unique redox properties of (H2O)2+• have been demonstrated by the transformation of benzene to phenol, C=C bonds to epoxides, and C=O bonds to generate [M + H2O]+•. Most recently, we discovered that (H2O)2+• can convert even such a highly inert molecule as nitrogen (N2) to yield nitroxyl (HNO) and hydroxylamine (NH2OH). The wide diversity of (H2O)2+• chemistry has been attributed to the co-existence of two distinct structures: [H3O+••••OH] and [H2O•••OH2]+•, which can exhibit the properties of •OH, H3O+, and H2O+•.
In our recent study (https://doi.org/10.1126/sciadv.adw0584) we demonstrated the potential of (H2O)2+• for the catalyst-free ambient conversion of CH4 to methanol with a remarkable yield of > 650 mmol·h–1 and the high conversion rate of approximately 35%. We showed that this surprisingly high conversion efficiency is attained owing to the peculiar one-step mechanism of this reaction: upon collision, (H2O)2+• cations effectively activate the highly inert methane molecule into its excited triplet state and then convert it into methanol – all within a single collision (see Figure 1).
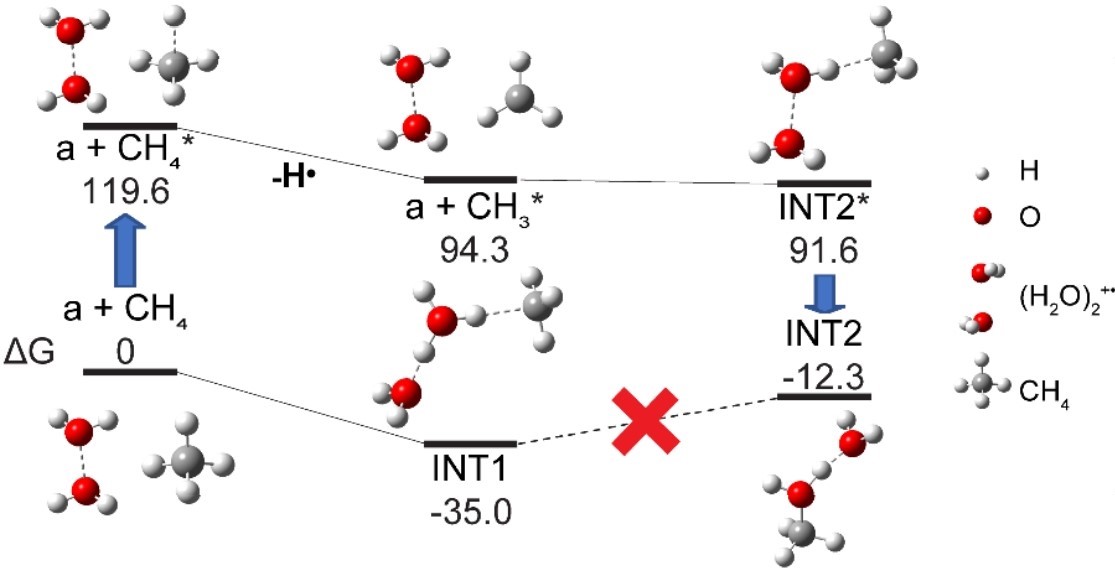
As a result, methane is selectively and controllably converted to methanol without cascade overoxidation, the sole by-product being hydrogen gas, produced at the rate of over 2 mmol·h–1. Compared to the previously reported catalytic methods, the production yields of our catalyst-free approach are up to 102~106 times higher for methanol and 103 times higher for hydrogen. This breakthrough opens the door to energy-efficient and cost-effective methods for the controllable conversion of methane without unwanted oxidation by-products such as CO2. These advancements could facilitate the adoption of methane in advanced industrial processes beyond combustion, thereby offering substantial environmental, economic, and ecological advantages.
Looking from a broader perspective, our results highlight the distinct possibility to implement “exotic” excited-state chemical transformations with industrially relevant yields. The experimentally manifested potency of (H2O)2+• to activate inert molecules, such as N2 and CH4, into their triplet states overcomes the severe thermodynamic and kinetic restrictions associated with ground-state transformations. This opens completely new synthetic spaces through utterly new reaction mechanisms with evident industrial prospects. The design ideas in this work should motivate more research efforts to further explore the distinct property of (H2O)2+• and similar systems to promote excited-state chemical transformations.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in