
Microbial communities can exhibit sophisticated phenotypes that do not pertain to any of the individual populations within them, but from their interactions. How can we see it? A collection of molecular tools, the “meta’omics techniques”, enabled microbiologists to study communities in vivo, bypassing often-arduous culturing campaigns. However, the meta’omics offer only a partial answer because they are not easily amendable to test for functional phenotypes in the lab and to address absolute quantification problems. In this study we addressed the latter via adaption and integration of absolute quantification methods for metatranscriptomics, metaproteomics and metabolomics from a time series experiment using a minimalistic cellulose-degrading and methane-producing consortium (SEM1b), resolved at the strain level with metagenomics and two strain isolates.
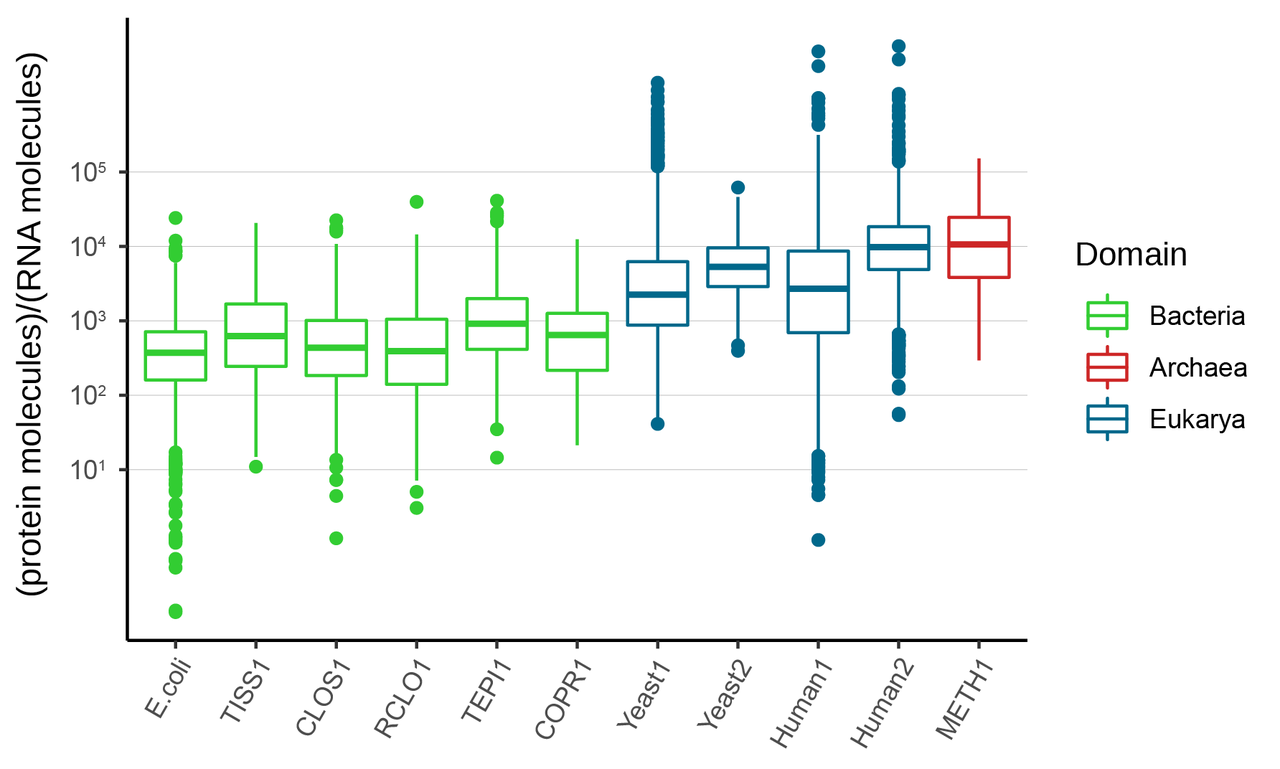
The protein-to-RNA ratio (during exponential growth phase) for the bacteria in SEM1b ranged ~102-104, corroborating the reference in literature (Escherichia coli); whilst the SEM1b’s archaeon, the methanogen Methanothermobacter thermautotrophicus, scored ~103-105, paralleling the Eukaryotic representatives yeast and human (Figure 1).
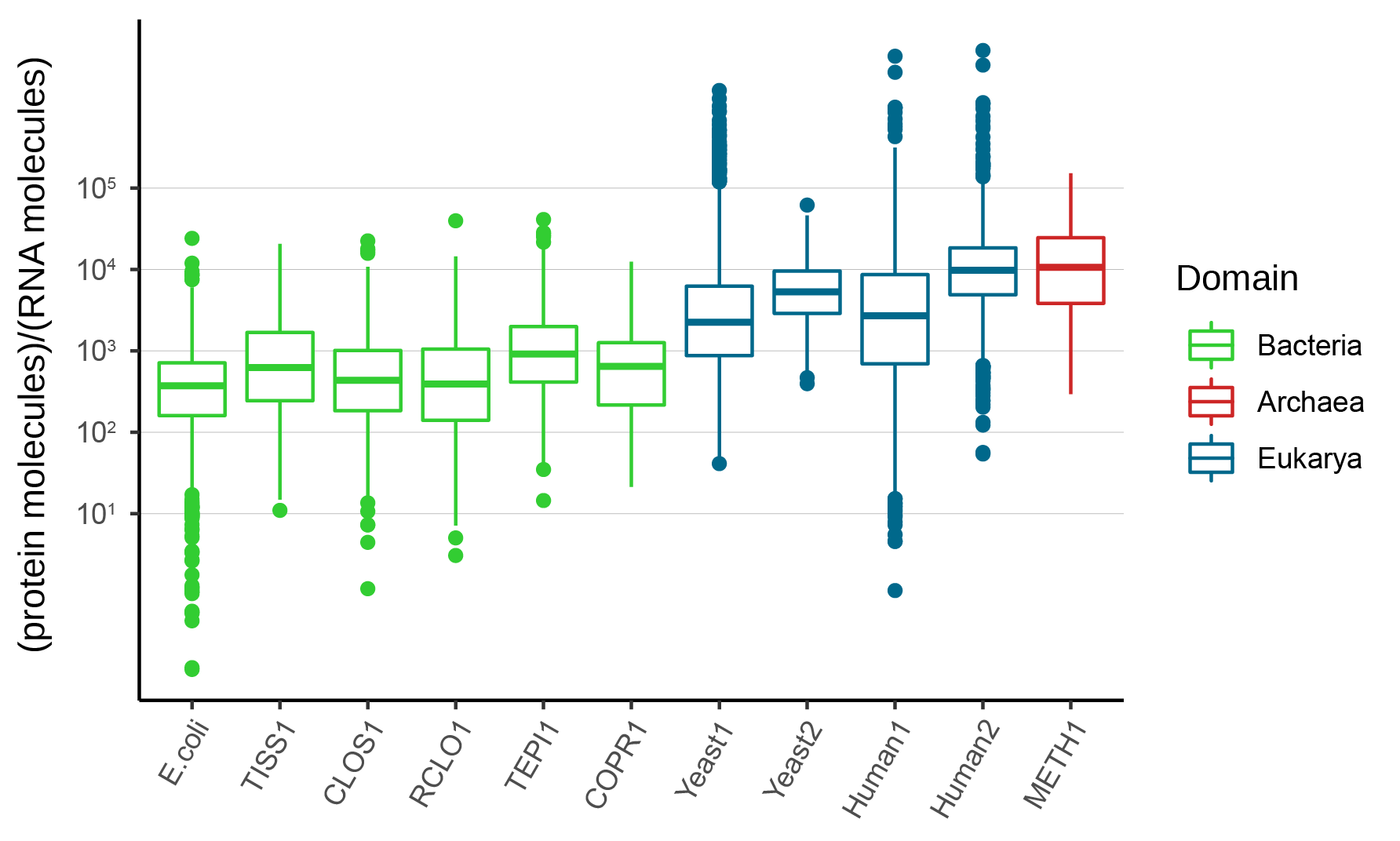
Figure 1. Protein-to-RNA ratios from representative organisms along the three domains of life. TISS1, CLOS1, RCLO1, TEPI and COPR1 are bacteria from SEM1b whilst METH1 is the archaeon of the community.
This evidence for Archaea joins the literature showing their many similar features with the Eukarya. Nonetheless, when we consider methanogens in natural environments (e.g. the cow and sheep rumen or biogas reactors) they are outnumbered by bacterial populations, often by an order of magnitude or more. Yet they perform the keystone function of maintaining H2 levels low enough to allow efficient fermentation and microbiome homeostasis. Could the ability of methanogens to maintain high protein levels at a lower RNA cost partially explain their entrenched niche in certain digestive ecosystems and the difficulties that other H2-consuming bacteria have competing with them?
We also tested the theoretical relationship between transcriptome and proteome in and out of steady-state, assumed during constant population growth in bacteria. We modelled the relationship with a monomial function and used the exponent k as proxy for the population status (k=1 indicates steady state). The temporal trajectories of the k-values per population were dynamic, with CLOS1, METH1 and TEPI1 populations converging to the same values, whereas RCLO1 had a parallel trend; suggesting that they possess a metabolic equilibrium (Figure 2a). Interestingly, these populations were the bulk “metabolic carriers” of SEM1b, with RCLO1 and CLOS1 degrading (hemi-)cellulose into short chain fatty acids, which were oxidised by TEP1 to produce H2+CO2/formate that were ultimately used by METH1 to generate methane (Figure 2b).
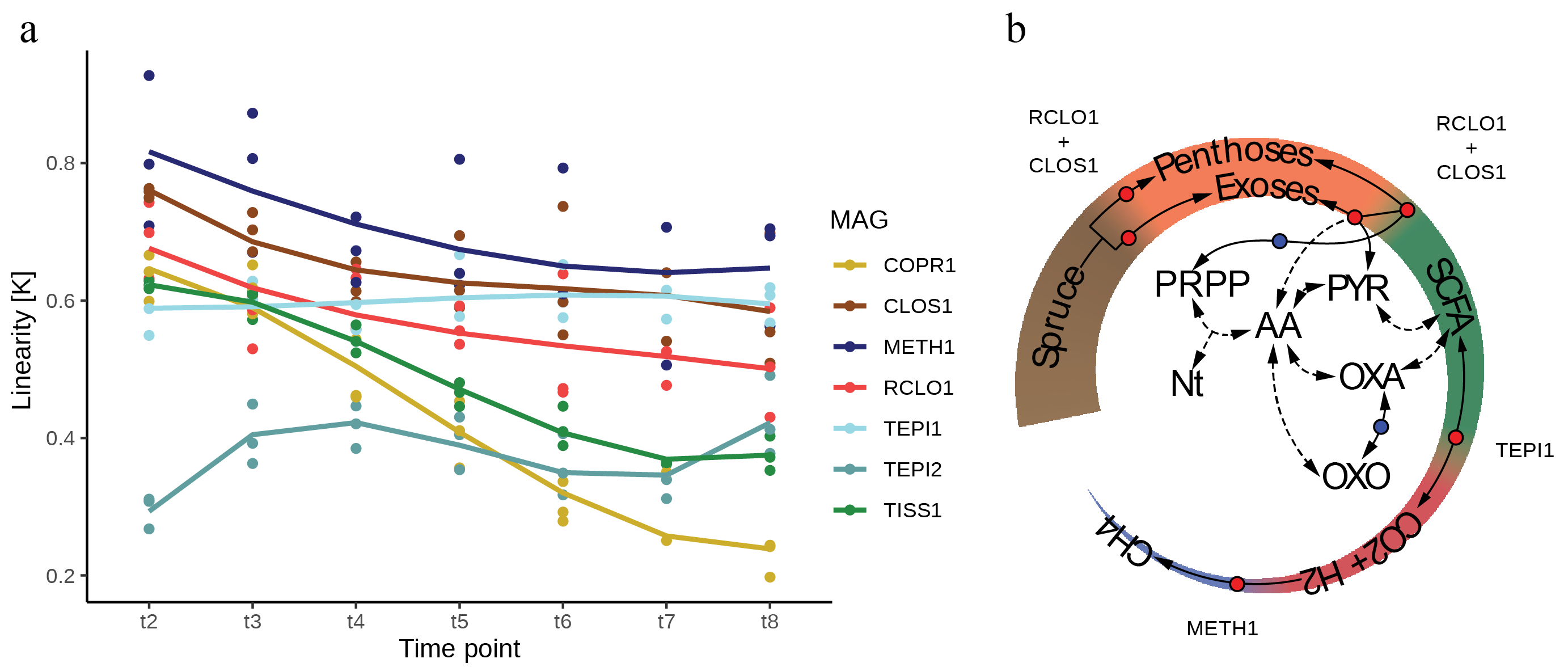
Figure 2. Dynamic k-values per population and metabolic summary chart of SEM1b.
Using this metabolic reconstruction and functional complementarity between these populations, we hypothesized that the relationship between transcriptome and proteome can highlight ecological cliques in complex microbiomes. However, this left us with open questions, such as: can a “k-status” be used to identify unknown synergies in more complex and less understood systems? Moreover, the estimation of change-points in the protein level regulation highlighted post-transcriptional regulation targets among the metabolic functions of the community carriers. These results and the dynamic k-value trajectories, hint to imagine that there are mechanisms in the prokaryotes to stir a population’s phenotype at the post-transcriptional level when confronting environmental challenges. The specific mechanisms may be tailored by evolution to fit strategies and niches of individual populations.
We are excited to see if our questions can be answered with larger and more detailed future experiments, and if these approaches can uncover completely novel forms of integrated systems spanning from gene regulation within individual populations to overarching community metabolisms!
Full article here.
Follow the Topic
-
Nature Communications

An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Advances in neurodegenerative diseases
Publishing Model: Hybrid
Deadline: Mar 24, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in