When Structure Persists: 3D Chromatin Memory Beyond Activity
Published in Protocols & Methods, Genetics & Genomics, and General & Internal Medicine
Gene regulation is often viewed as cell-type specific, dynamic, and tightly coupled to the presence of relevant transcription factors. Yet development leaves traces. Here, we ask whether chromatin architecture retains a form of memory, preserving regulatory relationships even in the absence of their functional output. We show that enhancer–promoter loops established during development can persist in differentiated progeny.
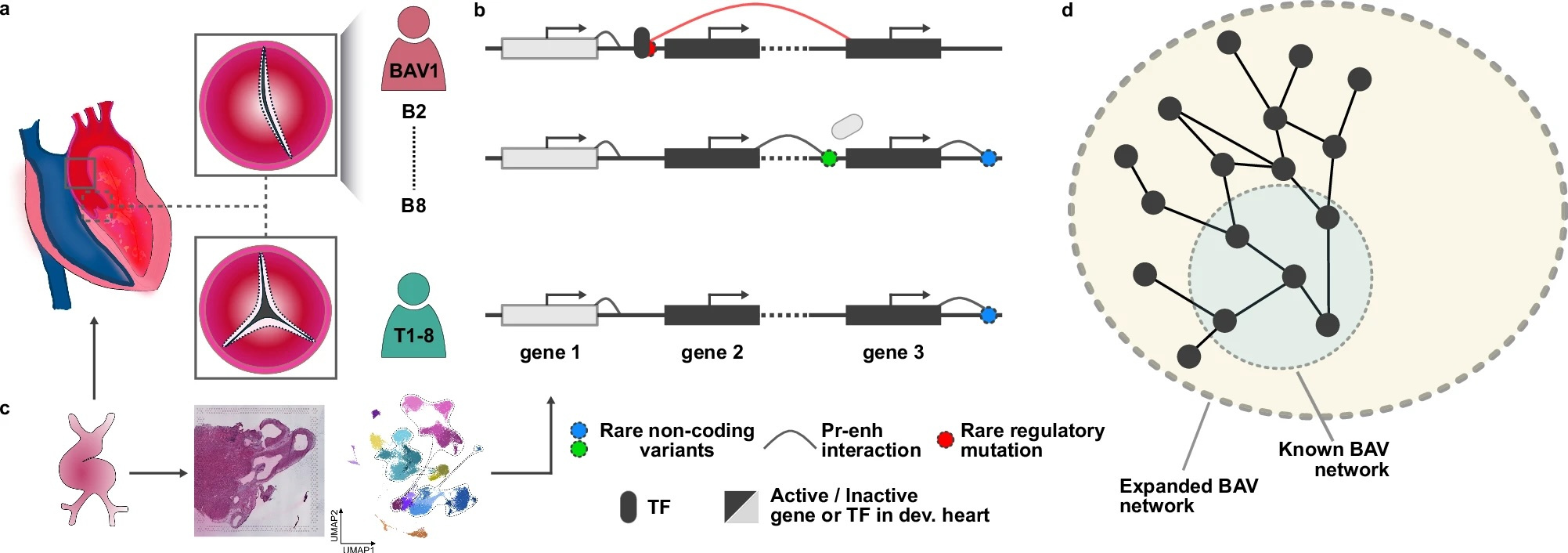
Many of these persistent loops are transcriptionally silent, likely because the necessary trans-acting factors are absent; as a result, enhancers are not actively driving gene expression. However, the physical proximity between enhancer and promoter remains encoded in the three-dimensional genome. This decoupling of structure and activity suggests that chromatin interaction maps capture not only current regulatory programs, but also a historical layer of genome organization shaped during development.
We leverage this concept of chromatin memory to address a longstanding challenge in human genetics—the identification of pathogenic regulatory mutations. Many disease-associated variants lie in non-coding regions, and linking them to their target genes remains difficult, particularly when the relevant regulatory interactions are inactive in the assayed cell type. By incorporating persistent enhancer–promoter loops into our analysis, we recover candidate regulatory relationships that would otherwise be missed.
Our results suggest that the regulatory landscape of adult cells extends beyond what is currently active. It also reflects prior developmental states, encoded in stable chromatin contacts. This latent regulatory layer provides a new framework for interpreting non-coding variation, enabling the discovery of mutations that act through developmental mechanisms but manifest in adult disease.
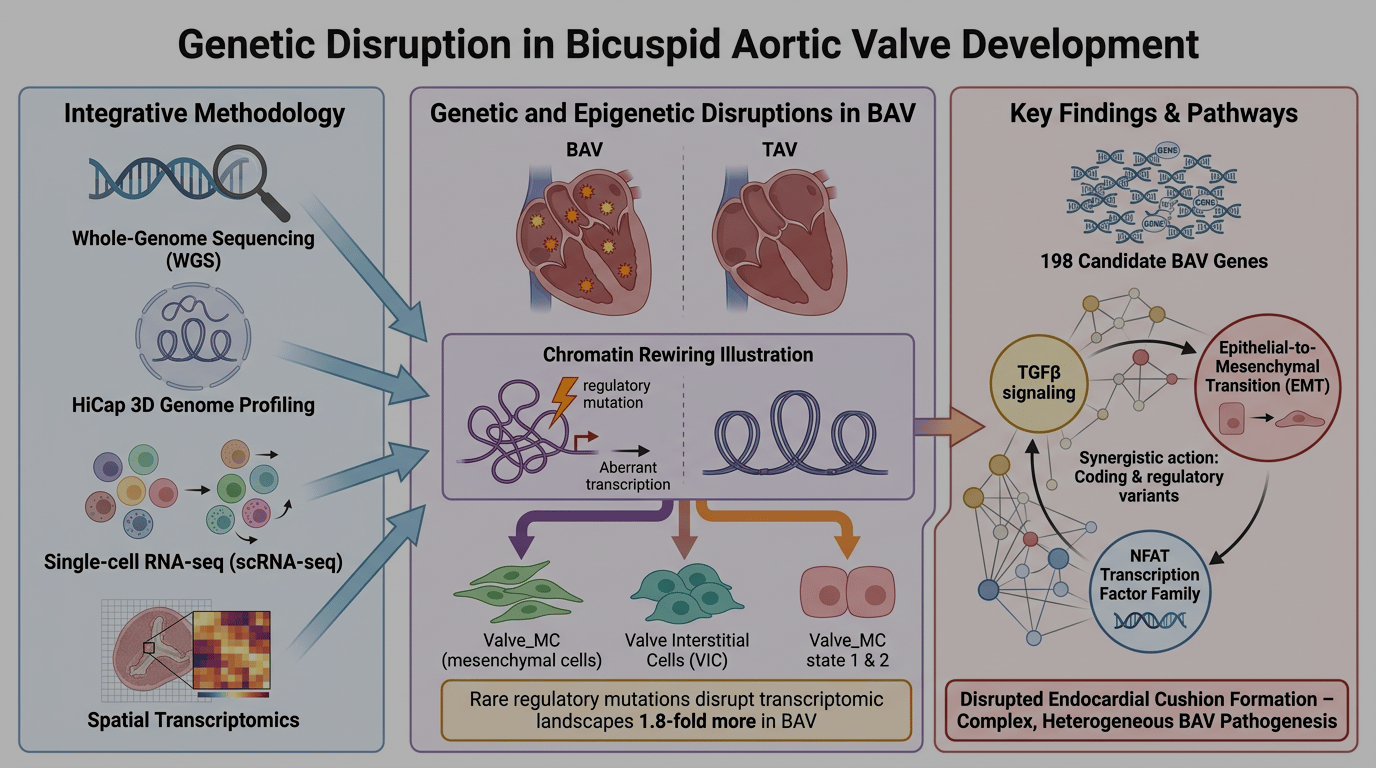
To demonstrate this, we use bicuspid aortic valve (BAV) as a model system—a congenital valve defect in which the aortic valve has two cusps instead of three. Its genetic etiology remains largely unresolved: only ~10% of cases can be explained by coding mutations, and genome-wide association studies have not identified common variants that account for the disease. We performed promoter-capture Hi-C (HiCap) on ascending aorta samples from 16 individuals, including eight patients with BAV. By integrating whole-genome sequencing and spatial transcriptomics data from fetal hearts with enhancer–promoter interaction maps, we show that the etiology of BAV is far more complex than previously appreciated. We identify tens of rare regulatory variants per patient that converge on a small set of genes with critical roles in valve development.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Jun 30, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in