A new dataset encompassing seven sequencing technologies on complex synthetic microbial communities
Published in Research Data

Metagenomics has revolutionized our understanding on the structure and composition of complex microbial communities, through whole shotgun sequencing on DNA directly extracted from the samples and also through the development of sophisticated bioinformatical software. To evaluate and validate bioinformatical tools for microbial metagenomics, mock samples with known composition are desired, yet it is hard to find complex synthetic microbial samples to validate tools and hypotheses and push them to their limits.
One of the primary goal of our paper published in Scientific Data was to generate DNA-based synthetic microbial communities, not obtained from in silico simulations, and complex enough to get closer to the diversity of natural biological communities. A few such synthetic communities are available from commercial sources but have a much lower diversity that the ones we constructed for this study. Our intent was not to mimic a community from a specific environment but to achieve a high degree of diversity at all phylogenetic levels and to capture a wide range of genomic sizes and composition (up to 87 species per mock community). We also focused on microbes that have reference completed genomes, although a few are draft genomes as we wanted to include those particular taxa. We decided to exclude a variety of microbes for which we had DNA but the genome sequence data was not adequate or we determined that there was some contamination present.

To assemble the synthetic communities, we used individually extracted DNAs from pure cultures of Archaea and Bacteria. Some strains and DNAs were obtained from various culture collections (DSMZ, ATCC), others from collaborators, others are isolates we obtained ourselves from various sources (human microbiome, various other environments). We grew many of the organisms in the lab (Oak Ridge National Laboratory), ranging from hyperthermophiles to aquatic, soil and human isolates and extracted the DNAs from those. It took time and effort, and for some it was linked to other culturomics projects. One of the most difficult part was to get the collection of DNAs spanning all the diversity we aimed for, including quantifying each and every DNA so that the relative abundance in the mock was as accurate as possible. Our synthetic communities ultimately included 91 species, some taxonomically closely related, spanning 29 archaeal and bacterial phyla.
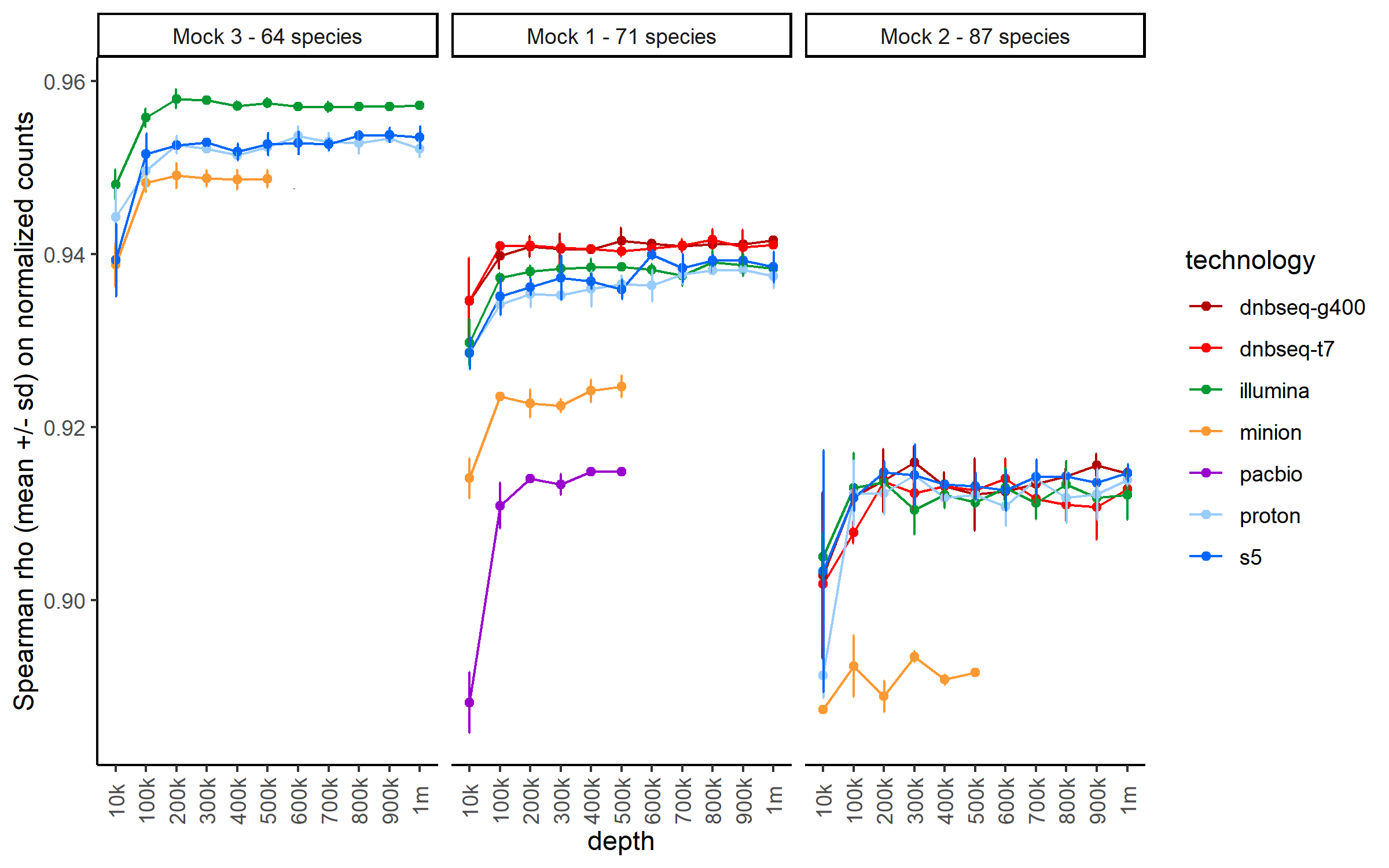
Beyond synthetic communities, we also wanted to capture the diversity of the different sequencing technologies, including second and third generation platforms. By the time our study began, we covered the most used and popular technologies for microbial metagenomics (Illumina HiSeq 3000, MGI DNBSEQ-G400 and DNBSEQ-T7, ThermoFisher Ion GeneStudio S5 and Ion Proton P1, Oxford Nanopore Technologies MinION R9 and Pacific Biosciences Sequel II). In particular, one of our question was to test and evaluate the performance of sequencing technologies in the context of quantitative metagenomic analysis, something that is usually missing in existing papers, where only performance of taxonomical composition is assessed.
At the beginning of the study, we were aware of the strong requirements for long read sequencing, for which long DNA fragments with high purity were required, a criterion that can be limiting when studying natural communities with low microbial cells concentration. Thus, we expected lower performance for long quantitative metagenomic analysis, compared to conventional short reads methods for microbial metagenomics. To our surprise, we measured very good performance for all technologies, even at a low sequencing depth (shallow sequencing), suggesting interesting opportunities to conduct whole shotgun sequencing on relatively low biomass microbial communities when considering only taxa exploration.
Of course, sequencing technologies are evolving rapidly and bioinformatics will need to further develop as well to effectively handle both short and long reads data, separately or together. This paper provides new resources to challenge assembly, taxonomy profiling and binning software as the synthetic communities produced in our study combine high complexity in number of species and include closely related organisms, a difficult study case for most of the existing software. As long read sequencing quality continues to rapidly improve, we believe that future metagenomic studies will soon incorporate both short and long reads as standard protocols.
Follow the Topic
-
Scientific Data
A peer-reviewed, open-access journal for descriptions of datasets, and research that advances the sharing and reuse of scientific data.

Related Collections
With Collections, you can get published faster and increase your visibility.
Computer vision in plant science and agriculture
Publishing Model: Open Access
Deadline: Oct 10, 2026
Datasets in education
Publishing Model: Open Access
Deadline: Nov 19, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in