Our paper entitled “Heterochiral coupling in non-ribosomal peptide macrolactamization” in Nature Catalysis can be found here: https://www.nature.com/article...
Penicillin-binding protein (PBP) is a key family of enzyme responsible for late-stage maturation and remodeling of bacterial peptidoglycan. They catalyze the formation or hydrolysis of an amide bond consisting of D-amino acid by forming an acyl-enzyme intermediate through a catalytic serine residue. PBPs are targets of well-established antibiotics – β-lactams – that structurally resemble their physiological substrate, D-Ala-D-Ala dipeptide motif in stem pentapeptide. To cope with this situation, bacteria have evolved a PBP-like enzyme, β-lactamase, that is specialized for hydrolyzing β-lactam rings.
Apart from the warfare at cell wall, we are now witnessing an increasing number of PBP-like enzymes being involved in other biological processes, such as secondary metabolism. Some PBP-like enzymes hydrolyze amide bonds consisting of D-amino acids (as in the case of peptidoglycan metabolism) to activate1, or de-activate2 the function of natural products. PBPs have fascinated the biocatalyst research community due to their enantiospecific amide bond-forming/hydrolyzing activities3. However, to the best of our knowledge, a PBP-like enzyme that is specialized for intramolecular amide bond formation (cyclization) had not been known, until the discovery of PBP-type thioesterase (TE) in 20184.
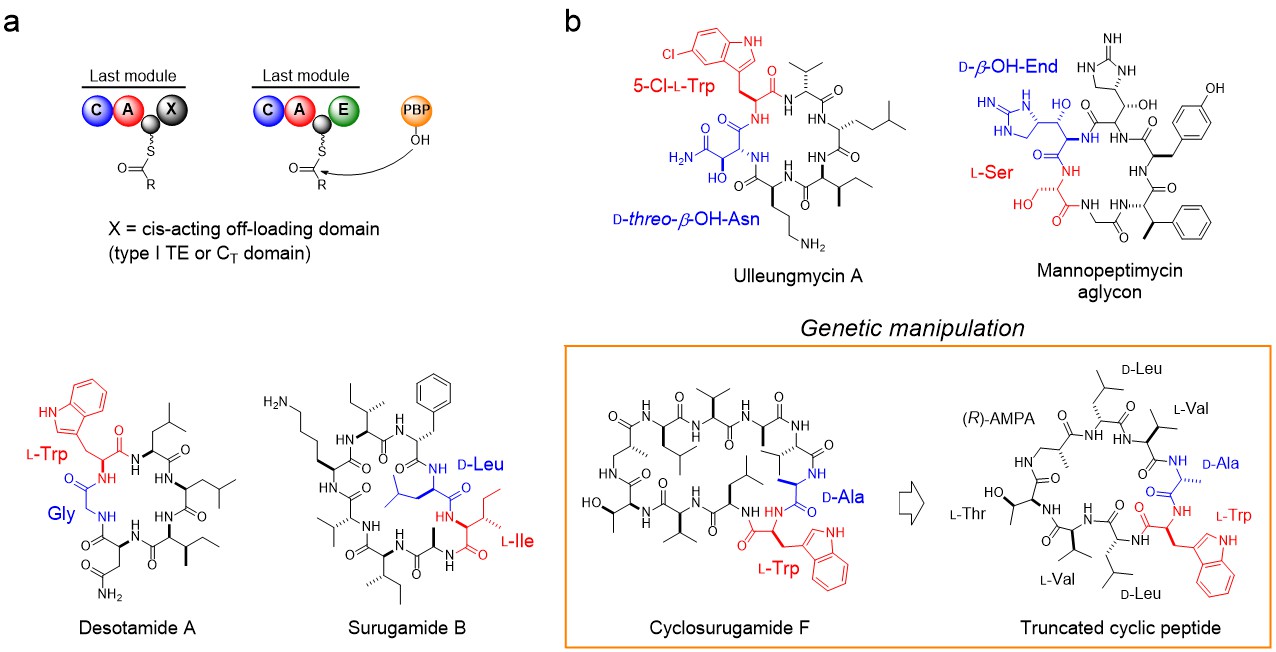
Figure. PBP-type TEs for non-ribosomal macrolactam biosynthesis. (a) Schematic comparison of the offloading steps mediated by canonical offloading domains and PBP-type TE. (b) Selected macrolactams biosynthesized by PBP-type TEs. The residues at the N and C termini of the linear precursors are coloured red and blue, respectively. Truncated assembly-line yielded a truncated cyclic peptide, which is designated as compound 13 in our paper.
SurE, the first member of PBP-type TE to be identified, is involved in biosynthesis of non-ribosomally synthesized macrolactams, surugamides. Surugamides are composed of two groups of cyclic peptides that differ in terms of size and amino acid sequence. Although they are synthesized by two distinct non-ribosomal peptide synthetases (NRPSs), they are both cyclized and released from NRPSs by a single promiscuous cyclase, SurE, which doesn’t show sequence homology to canonical offloading domains but is rather similar to PBPs4,5.
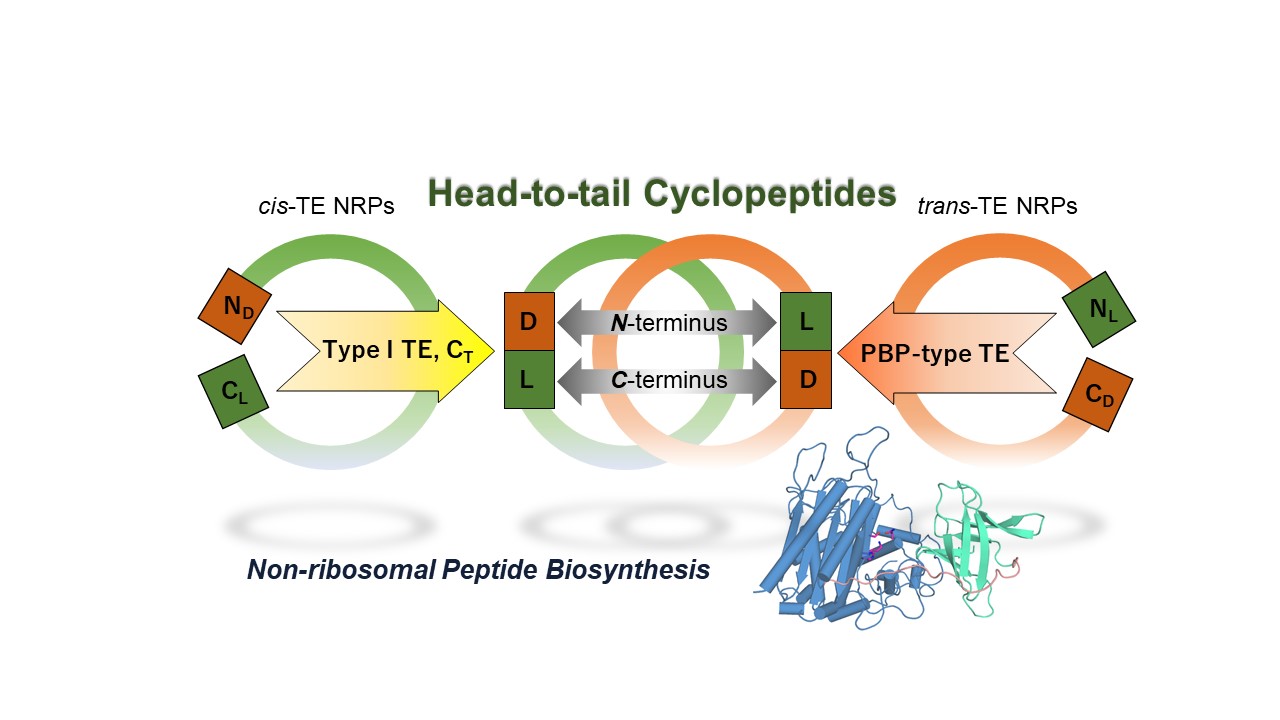
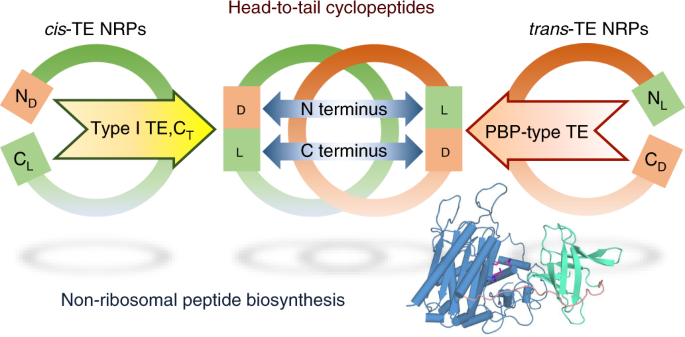
As chemo-, and regio-selective cyclization of peptide are of interest but remain challenging tasks6, in our paper published on Nature Catalysis, we investigated the scope and limitation of this intriguing peptide cyclase with an array of synthetic substrate analogs. This revealed that – while SurE tolerates substitutions of residues in the middle of the substrate – it requires heterochirality between N- and C-terminal residue with L-, and D-amino acid, respectively. It is worth noting that the peptide bond formation exhibits significant preference for heterochiral coupling also in abiotic condition7. This fact prompted us to examine the enantioselectivities of other NRP cyclases. From our survey on domain architectures of NRPSs for head-to-tail macrolactams, it turned out that the canonical cis-acting offloading cyclases, such as the type I TE and terminal condensation-like (CT) domains, also couple heterochiral residues, but with inverse stereoselectivity to PBP-type TE. Therefore, it seems that heterochiral coupling is a common trait of biosyntheses of non-ribosomal macrolactams and that bacterial NRPS recruited D-amino acid-recognizing PBP-like enzyme to overcome the sterochemical constraints present in the heterochiral head-to-tail macrolactamization. Our first X-ray crystal structure on this family of cyclase provided structural rationale for its C-terminal stereospecificity.
We next questioned whether SurE exclusively cyclize a peptide chain tethered on the last module of assembly-line. To test this, we simply deleted the last two modules at the C-terminus of the decapeptide synthetase so that the truncated assembly-line would synthesize a shortened peptide with D-amino acid at its C-terminus. It was interesting to find that this manipulated assembly-line actively produce the truncated version of cyclopeptide, indicating that SurE could cyclize a non-natural peptidyl substrate tethered on a non-cognate peptidyl-carrier protein.
Overall, this study shed light on an unusual stereochemical requirement of PBP-type TE. Also, SurE performed broad substrate specificity both in vitro and vivo, highlighting its potential as a biocatalyst for peptide cyclization and as a tool for synthetic biology. These results set the stage for further mechanistic analysis and investigation on catalytic capabilities of this valuable family of enzyme.
[1] (a) Brotherton, C.A. & Balskus, E.P. J. Am. Chem. Soc. 135, 3359-3362 (2013). (b) Reimer, D. & Bode., H.B. Nat. Prod. Rep. 31, 154–159 (2014).
[2] Li, Y. et al. Nat. Chem. Biol. 14, 381–387 (2018).
[3] (a) Asano, Y. et al. Angew. Chem., Int. Ed. 28, 450–451 (1989). (b) Wehofsky, N. et al. Angew. Chem., Int. Ed. 47, 5456-5460 (2008).
[4] Kuranaga, T. et al. Angew. Chem. Int. Ed. 57, 9447–9451 (2018).
[5] Matsuda, K. et al. Org. Biomol. Chem. 17, 1058–1061 (2019).
[6] (a) White, C. J. & Yudin, A. K. Nat. Chem. 3, 509–524 (2011). (b) Yu, X. & Sun, D. Molecules 18, 6230–6268 (2013).
[7] (a) Hill, R. R., Birch, D., Jeff, G. E. & North, M. Org. Biomol. Chem. 1, 965–972 (2003). (b) Brady, S. F. et al. J. Org. Chem. 44, 3101–3105 (1979). (c) Rich, D. H., Bhatnagar, P., Mathiaparanam, P., Grant, J. A. & Tam, J. P. J. Org. Chem. 43, 296–302 (1978).
Follow the Topic
-
Nature Catalysis
This journal brings together researchers from across all chemistry and related fields, publishing work on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, incorporating both fundamental and applied studies.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in