Acetyl-CoA influences the secretory pathway and neuronal activity
Published in Neuroscience and Anatomy & Physiology
INTRACELLULAR ACETYL-COA FLUX MODULATES PROTEIN QUALITY CONTROL
Cellular homeostasis depends on precise crosstalk among organelles and compartments, mediated by sensing and signaling pathways that integrate both extracellular and intracellular inputs. Central to these adaptive mechanisms are metabolites that act not only as enzymatic substrates but also as potent signaling regulators, influencing enzyme activity, gene expression, and protein modification.
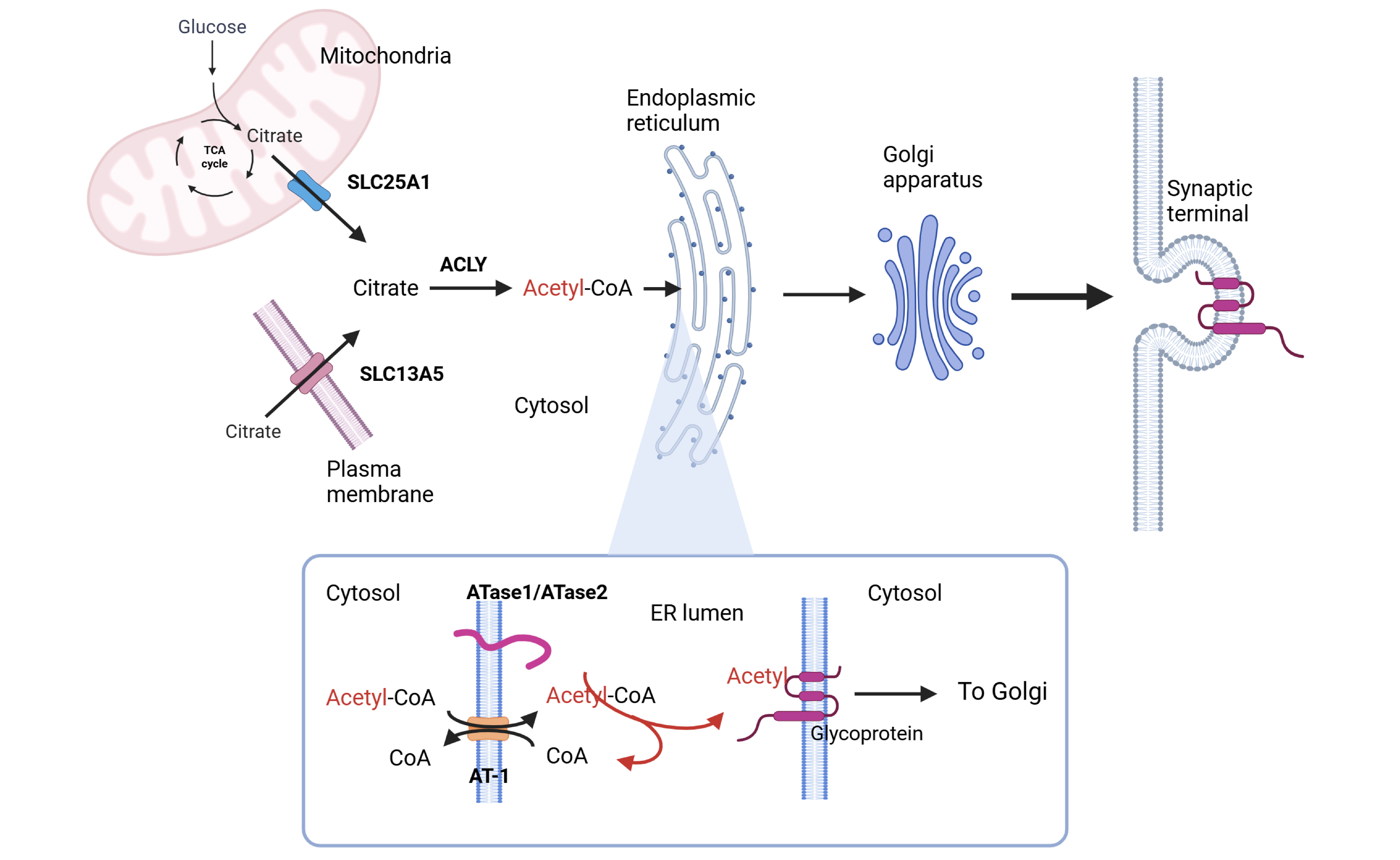
Among these metabolites, acetyl-CoA has emerged as a central node in nutrient signaling. Dynamic fluctuations in its intracellular flux provide direct substrate-level control over protein activity and different metabolic pathways, allowing rapid reprogramming of cellular functions in response to metabolic demands. By linking metabolic state to the functional adaptation of the secretory pathway, ATase1 and ATase2 are now recognized as novel components of the nutrient-signaling network (PMCID: PMC9792894). These two enzymes are responsible for Nε-lysine acetylation in the lumen of the endoplasmic reticulum (ER), targeting both ER-resident and ER-cargo proteins (PMCID: PMC6262770).
ER acetylation has emerged as a key branch of the ER quality-control machinery. Correctly folded polypeptides are acetylated while unfolded/misfolded polypeptides are not. The former are allowed to transit from the ER to the Golgi apparatus while the latter are not.
The activity of ATase1 and ATase2 is allosterically regulated by acetyl-CoA and CoA availability in the ER lumen. Specifically, acetyl-CoA activates ATase activity, whereas free CoA inhibits it. The acetyl-CoA/free CoA gradient across the ER membrane is maintained by AT-1/SLC33A1, an ER membrane transporter that exchanges cytosolic acetyl-CoA for ER luminal CoA. AT-1 operates downstream of the SLC25A1 and SLC13A5 network. SLC25A1, a mitochondrial membrane transporter, delivers citrate from mitochondria to the cytosol, while SLC13A5, a plasma membrane transporter, imports citrate from the extracellular space. In the cytosol, citrate is converted by ATP-citrate lyase (ACLY) into acetyl-CoA, thereby fueling ER acetylation.
In summary, the citrate/acetyl-CoA/ER acetylation pathway plays a critical role in quality control of ER-synthesized proteins and consequently of many cellular functions mediated by ER-synthesized proteins.
ACETYL-COA FLUX AND AUTISM SPECTRUM DISORDER WITH INTELLECTUAL DISABILITY
The brain represents about 2% of total body mass but consumes about 20% of the body’s energy. This substantial energy demand primarily supports presynaptic and postsynaptic activities. Neurons, in particular, depend heavily on the secretory pathway to ensure efficient delivery of properly folded polypeptides to synaptic terminals. Proper functioning of this pathway is therefore particularly critical in neurons.
Gene duplication events affecting 22q11.21 (harboring SLC25A1), 17p13.1 (harboring SLC13A5), 3q25.31 (harboring AT-1), and 2p13.1 (harboring ATase1 and ATase2) are all associated with developmental delay, autism spectrum disorder (ASD) with intellectual disability and progeria-like dysmorphism. These findings suggest that hyperactivation of the biochemical pathway that links citrate and acetyl-CoA availability to ER acetylation contributes to the behavioral alterations characteristic of certain rare forms of ASD.
A key limitation, however, is that the above gene duplication events often involve multiple genes. To establish direct causality between specific genes and disease phenotypes, mouse modeling is essential. With this goal in mind, we targeted each component of the ER acetylation machinery (AT-1, ATase1, and ATase2) along with SLC25A1 and SLC13A5 in the mouse.
HYPERACTIVITY OF THE ER ACETYLATION MACHINERY CAUSES AUTISTIC-LIKE FEATURES IN THE MOUSE
Mice with neuron-specific overexpression of the genes described above exhibit a consistent ASD-like phenotype across models (PMCID: PMC4925020; PMCID: PMC9014753; PMCID: PMC8823335). This phenotype includes ASD-like behavioral alterations, expansion of the somatodendritic compartment, increased synaptic connectivity, and neuronal hyperactivity. Together, these findings support the conclusion that the citrate/acetate/ER acetylation pathway plays a central role in ASD pathophysiology. Importantly, all experiments were conducted in mice of the same genetic background (C57BL/6J) using an identical expression system (Camk2a-tTA:TRE), enabling direct and reliable cross-model comparisons.
OVEREXPRESSION OF ATASE1 OR ATASE2 CAUSES AN ASD-LIKE PHENOTYPE
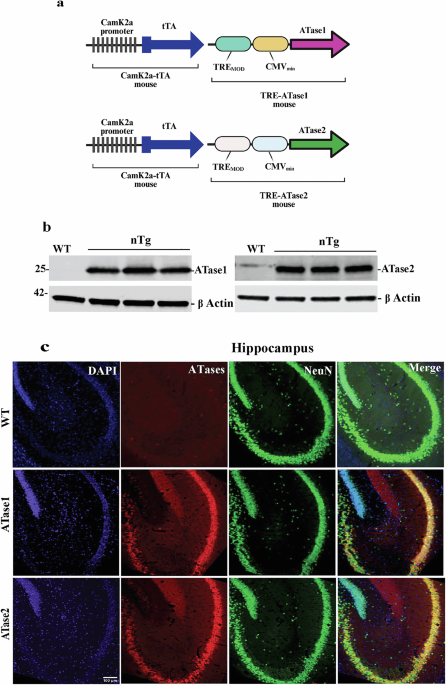
ATase1 and ATase2 are the last output of the biochemical pathway that links citrate and acetyl-CoA availability to ER acetylation. To study how the ATases influence the development of ASD, we generated C57BL/6J mice overexpressing either ATase1 or ATase2 in forebrain neurons (DOI: 10.1038/s41380-025-03228-1).
Behavioral testing across multiple standardized platforms revealed that ATase1- and ATase2-overexpressing mice displayed alterations comparable to those observed in other established ASD models. Neurons isolated from these mice exhibited abnormal morphology, including an increased number of synaptic connections. These structural changes were accompanied by heightened spontaneous neuronal activity, as measured with multielectrode arrays, and by altered synaptic plasticity, as determined through brain slice electrophysiology.
Multielectrode arrays record neuronal activity in isolated cultures, while brain slice electrophysiology measures activity within intact neuronal networks. Both approaches converged on the conclusion that ATase overexpression perturbs neuronal excitability and plasticity.
In conclusion, neuronal overexpression of ATase1 or ATase2 caused developmental and functional defects leading to an ASD-like phenotype. These genetically engineered models recapitulate the genetic association between the 2p13.1 duplication and the occurrence of developmental delay, ASD, and intellectual disability in humans. Additionally, when the ATases were overexpressed in the whole body (not just forebrain neurons), the mice developed a progeria-like phenotype (PMCID: PMC12423556).
OVEREXPRESSION OF ATASE1 OR ATASE2 ALTERS THE SECRETORY PATHWAY
Unbiased large-scale analysis of the brain proteome revealed defective engagement and efficiency of the secretory pathway with increased translocation of nascent glycoproteins to the neuronal surface in both mouse models. These defects impacted basic neuronal events involved in development, outgrowth of dendritic branches and formation of functional synapses.
BEYOND THE STUDY
The ASD associated with gene duplication of 22q11.21, 17p13.1, 3q25.31, and 2p13.1 are currently classified as separate rare diseases (conditions affecting fewer than 1 in 200,000 individuals). However, the above mouse models indicate that the same metabolic pathway -the CoA/acetyl-CoA pathway- is implicated in all of them. This suggests the possibility, requiring further study, that one common treatment might be useful for therapeutic purposes.
Furthermore, these ASD diseases account for only a small fraction of diagnosed autism cases. Although metabolomic studies of individuals with non-syndromic ASD have revealed converging metabolic alterations, follow-up studies will be required to assess whether the results obtained with the above mice targeting the CoA/acetyl-CoA pathway might be extended to non-syndromic forms of ASD.
Follow the Topic
-
Molecular Psychiatry
This journal publishes work aimed at elucidating biological mechanisms underlying psychiatric disorders and their treatment, with emphasis on studies at the interface of pre-clinical and clinical research.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in