AIMD-Chig: Exploring the conformational space of proteins at DFT level
Published in Bioengineering & Biotechnology

The landscape of life sciences research is being transformed as the precision of molecular dynamics simulations becomes virtually indistinguishable from that of wet-lab experiments. Molecular Dynamics (MD) simulations vary in the methods used to calculate forces. Classical molecular dynamics enables rapid simulations through the application of a predetermined interatomic potential function, while ab initio molecular dynamics utilizes potential derived from the electronic structure of molecules, providing accurate molecular characterizations but facing the challenge of excessive computational overheads for large systems. Recently, machine learning force fields (MLFFs) have shed light on balancing the accuracy and efficiency by fitting an inter-atomic potential while the high-quality data at ab initio accuracy is a prerequisite.
Several MD datasets have been generated at the Density Functional Theory (DFT) level. However, the computational cost increases cubically with the number of atoms, these datasets primarily focus on small organic molecules and have a limited variety of conformations. Our goal is to generate a comprehensive, full-atom protein MD dataset that explores the nearly entire conformational space. This will enhance the applicability of MLFFs for protein dynamics and enable the detection of protein behaviors that classical MD simulations cannot achieve.
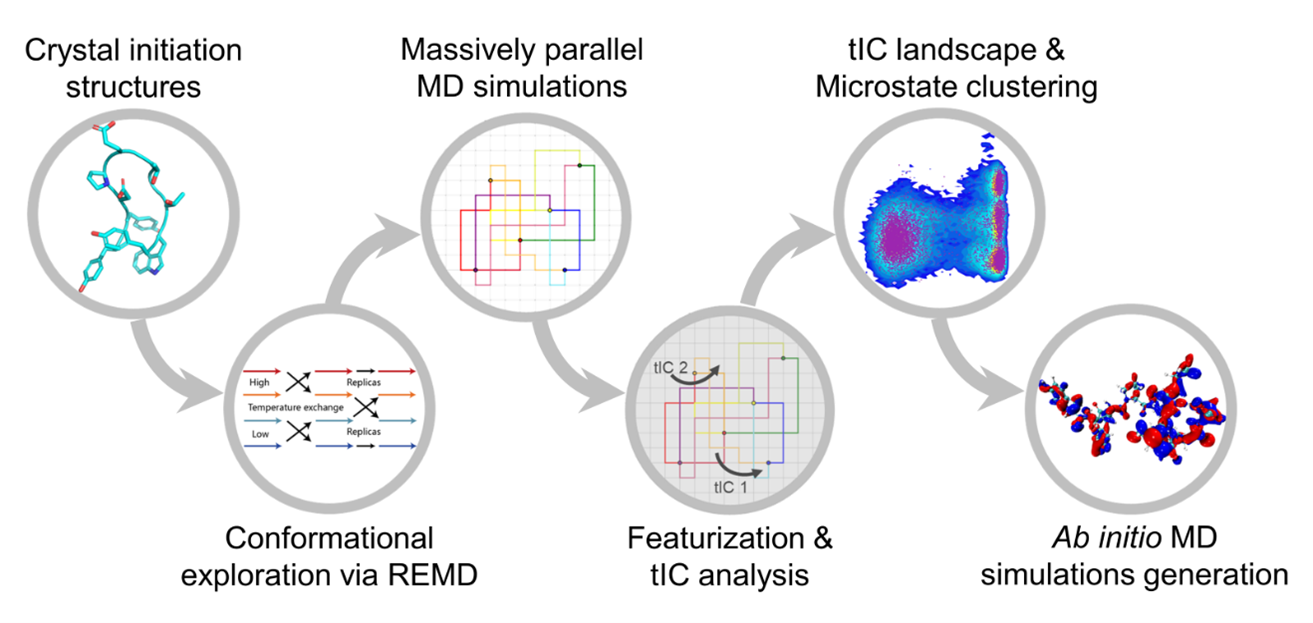
In this study, we focus on the simplest protein, Chignolin, which has only 166 atoms. The key challenge is how to fully explore the conformational space of proteins at the DFT level. According to our preliminary experiments, simulating Chignolin to a microsecond will take more than 28,000 years by quantum simulation. Thus, we design a novel technology by combining a series of classic MD simulations and quantum MD simulations and reduce the computational time from 28,000 years to 3 months. As shown in the poster image, we employ replica exchange MD and conventional MD to comprehensively explore the conformational space of Chignolin. Then we select the representative structures from massive simulation trajectories as "anchors" of the protein. The concept of "anchor" is key to our data generation technology. They are derived from fast classic MD simulations, represent the potential energy surface of the protein and are then fed into quantum simulation for accurate energy and force calculation. By taking the advantages from both sides, anchors connect between classic MD and quantum MD and complete the task with acceptable time consumption. As a result, 2 million samples are generated by employing over 7 million CPU core hours in 3 months.
An interesting thing is to utilize the dataset to train MLFF and run MD simulations driven by the model to gain new insights into protein dynamics. During the model training, we have found that the more data is incorporated, the better the model’s performance. Furthermore, when performing MD simulations powered by the model, a protonation process has been detected when a hydrogen bonded to a nitrogen is attracted by an oxygen atom in another residue and form a steady bond. This can serve as a starting point to study the phenomenon that classic MD cannot do. By utilizing the dataset (AIMD-Chig: exploring the conformational space of 166-atom protein Chignolin with ab initio molecular dynamics (figshare.com)), a large variety of research can be conducted to study the protein dynamics with ab initio accuracy.
This dataset is part of the outcomes of a larger project known as AI-powered Ab Initio Molecular Dynamics (AI2BMD) (AI2BMD: efficient characterization of protein dynamics with ab initio accuracy | bioRxiv), which makes use of AI to do fast molecular dynamics simulation for large molecular systems with near ab initio accuracy. It achieves near ab initio accuracy for energy and force calculations of proteins containing over 10,000 atoms. With the ability of simulating protein dynamics at ab initio accuracy, the project effectively complements laboratory experiments in understanding the dynamic aspects of various biochemical processes.
Dr. Tong Wang is the Senior Researcher at Microsoft Research AI4Science. He received his Ph.D at Tsinghua University and held a visiting Ph.D experience at Harvard University. His research focuses on algorithm design and applications on molecular dynamics simulation with artificial intelligence, machine learning force field, computer-aided drug discovery and protein structure prediction. Dr. Wang published a series of peer-reviewed papers as the first and corresponding authors on Nature, Nature Machine Intelligence, Nature Communications, Cell Research and so on, and holds several China and US patents. He is also the reviewers of Nature series journals and the honored reviewer of ACS publications. As the project lead, he proposed AI2BMD simulation program that transforms protein dynamics simulation with ab initio accuracy. As the team leader, he won the championship in the First Global AI Drug Development Competition (https://aistudio.baidu.com/competition/detail/1012/0/leaderboard) and the winner of OGB Large Scale Challenge@NIPS2022 competition. His research “Exploring the Regulatory Function of the N-terminal Domain of SARS-CoV-2 Spike Protein through Molecular Dynamics Simulation” won Top Downloaded Article Award by Wiley Press. He is the committee member of China Bioinformatics Society.
Follow the Topic
-
Scientific Data
A peer-reviewed, open-access journal for descriptions of datasets, and research that advances the sharing and reuse of scientific data.

Related Collections
With Collections, you can get published faster and increase your visibility.
Computer vision in plant science and agriculture
Publishing Model: Open Access
Deadline: Oct 10, 2026
Wearable and Computer Vision Data for Health and Behaviour Research
Publishing Model: Open Access
Deadline: Aug 08, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in