Arabidopsis mass spectral library: a resource for plant proteomics
Published in Research Data
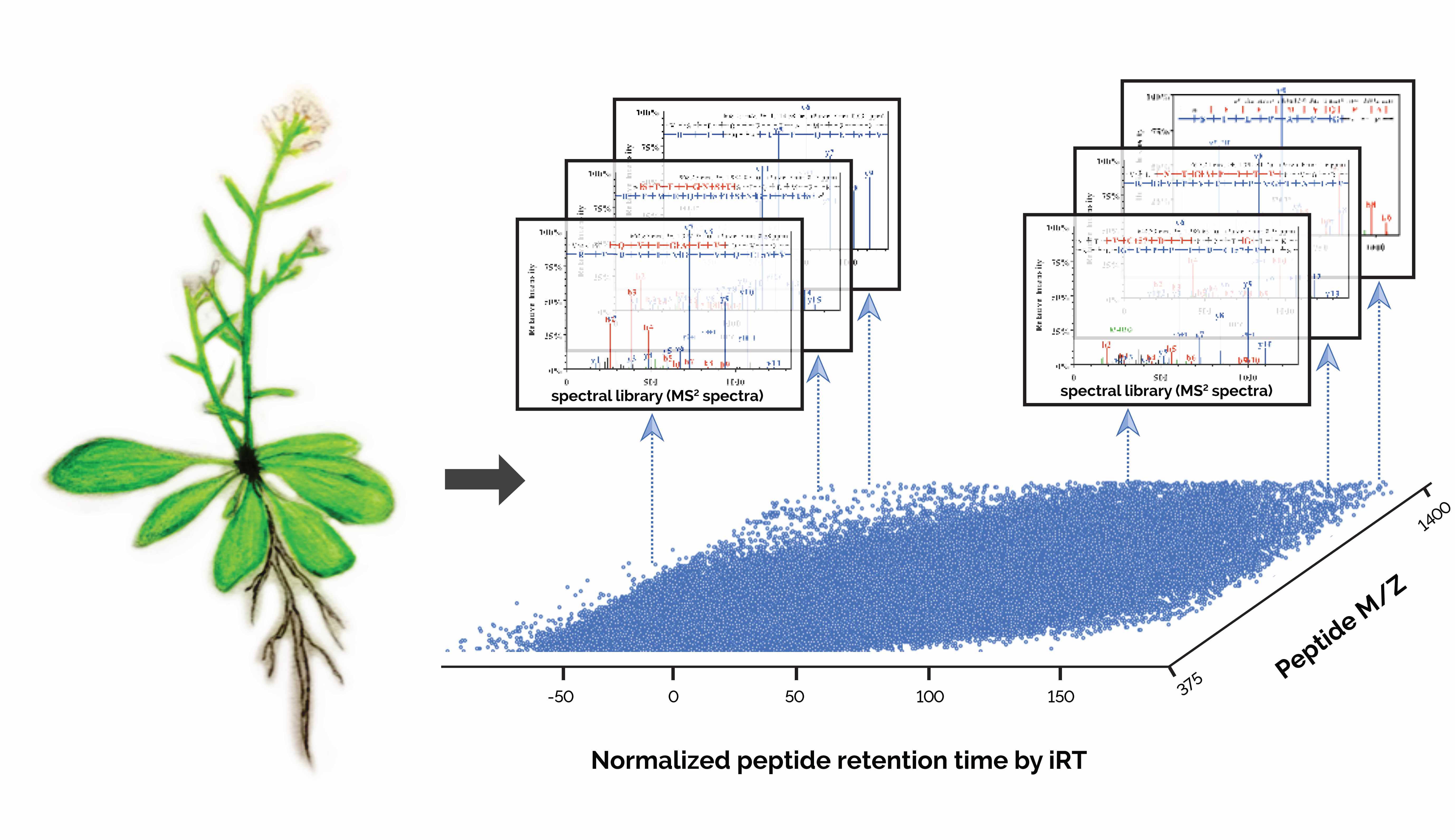
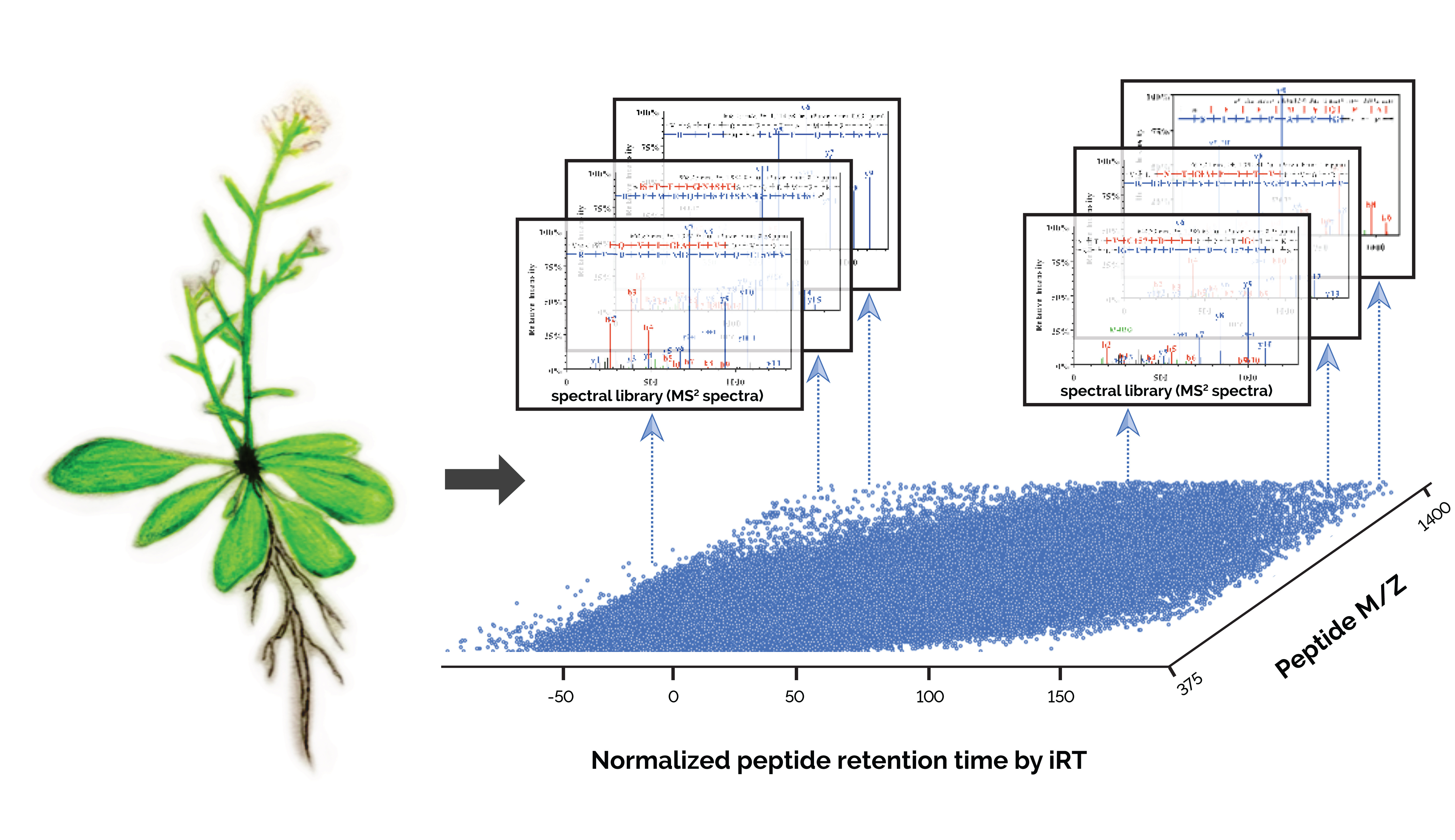
Arabidopsis is a small flowering plant with a short but complex life cycle. It exhibits high similarity to many crop plants in biology and physiology, and thus study of this species has important applications for agriculture. Since 1980s, Arabidopsis has become a widely used plant organism for laboratory research. In 2000, it became the first plant with its genome completely sequenced. Subsequently, genomics studies have greatly facilitated functional annotation of novel genes and genetic variations. In comparison, proteomics study of Arabidopsis lags far behind although proteins are generally considered with higher relevance of physiology. This was mainly due to the challenges that proteomics faces such as relatively lower coverage, quantitation accuracy and reproducibility.
In 2014, I attended a targeted proteomics workshop named “SRM Course”. During the course, Professor Ruedi Aebersold, a leading proteomics researcher, introduced us their newly development in SWATH-MS (Sequential Windowed Acquisition of All Theoretical Fragment Ion Mass Spectra) and the successful application to human biomarker discovery. This DIA-MS method is considered as a high-throughput, massively parallel targeted approach for accurate proteome quantification. In the approach, a comprehensive high-quality spectral assay library was constructed using conventional data dependent acquisition (DDA), which catalogs spectral of each detectable peptide within a given study material together with its precise retention time from liquid chromatography. Subsequent match of the optimal DIA-MS data to the library could give reproducible and accurate proteome quantitation.
I was so excited about this technique and eager to set up similar workflow in our proteomics core facility. We decided to use Arabidopsis as study material because of two considerations: 1) desert agriculture is one of key research areas in our university where many researchers use Arabidopsis for their research; 2) in proteomics community, many interests focus on mammals such as human and mouse. Thus, use of Arabidopsis could likely avoid redundant efforts from other researchers.
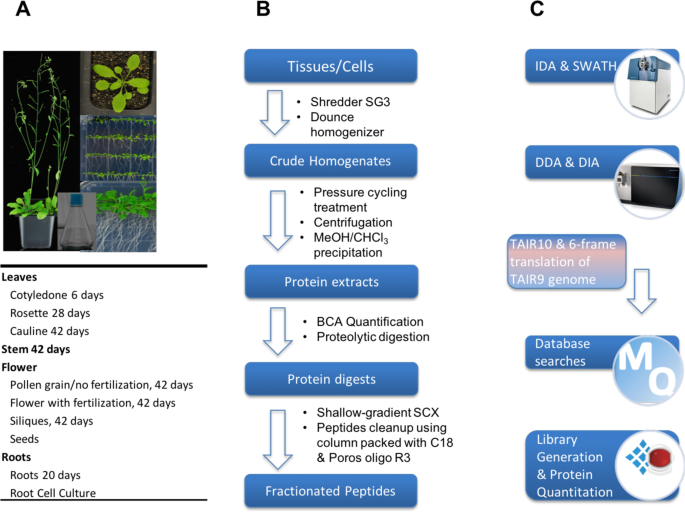
During the data collection, we encountered two big challenges. The first challenge was related to sensitivity degradation in the MS instrument, and the difficulty in getting experienced engineer for the quadrupole cleaning. In this study, we analysed a total of 10 organs of Arabidopsis, with each organ containing about 30 injections. The MS performance dropped significantly due to space charge effect of quadrupole after running a large number of samples. However, the KAUST is young university in Saudi Arabia. At the early stage, getting support from outside including engineer visit, shipping chemicals and reagent were rather slow. This significant delayed the progress of our project. The second challenge was to obtain high coverage of protein quantitation. Despite having a high-quality spectral library containing over 15,000 protein groups, initial DIA-MS analysis quantified a relatively low number protein groups (approximately 3,000) from each sample. We reasoned that this was mainly due to the large dynamic range of Arabidopsis proteome and non-protein interference such as plant pigment. For example, the protein RuBisCo alone accounts for >50% of protein content in most of Arabidopsis organs. With subsequent improvements in both sample preparation and DIA-MS method, a significant higher number of proteins were quantified from our DIA-MS approach, which is comparable to DDA-MS analysis.
In the paper published today (https://www.nature.com/articles/s41597-019-0294-0), it presents the first comprehensive Arabidopsis mass spectral assay libraries obtained using two types of high-resolution mass spectrometry platforms. It details the methodology for the library generation and demonstrates the successful application of DIA-MS in the quantitation of Arabidopsis proteome variations in response to abscisic acid stress treatment. We expect that researchers will find this resource and the described DIA-MS approach useful for fast and reliably quantifying proteome variations under different conditions.
Follow the Topic
-
Scientific Data
A peer-reviewed, open-access journal for descriptions of datasets, and research that advances the sharing and reuse of scientific data.

Related Collections
With Collections, you can get published faster and increase your visibility.
Computer vision in plant science and agriculture
Publishing Model: Open Access
Deadline: Oct 10, 2026
Wearable and Computer Vision Data for Health and Behaviour Research
Publishing Model: Open Access
Deadline: Aug 08, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in