Beyond "dysbiosis": disease-specific and shared microbiome responses to disease
Published in Microbiology
The paper in Nature Communications is here: http://go.nature.com/2AcJvLY
When I joined Eric Alm's lab three years ago, I was energized by the excitement and activity in microbiome research, but frustrated with how little of the information in our field had been synthesized into generalized understanding. When I read new papers that came out, I wanted to know how much I could trust the results of any individual study: were the bacterial associations that they reported actually meaningful biomarkers for that disease, or were they just spurious associations that had simply passed statistical significance tests? I also didn't really recognize any of the bacterial names yet back then, but I felt like many of the same taxa kept showing up across multiple studies - were there bacteria that just popped up no matter what disease was being studied? And, finally, what the heck did people mean when they said that they had identified "dysbiosis"? It seemed to me that often, "dysbiosis" was just another way to say "difference" - but what kind of difference, and was it biologically meaningful?

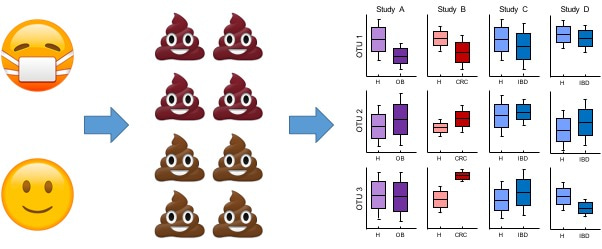
Motivated by these thoughts, I set out to do a broad cross-disease meta-analysis to see if I could "find the knowledge in the information." I wanted to answer two main questions: (1) were microbial associations in a given study specific to that disease, consistently showing up across different studies of the same disease, and (2) were there general health- and disease-associated bacteria, that were significant no matter what the studied disease was? Since our field is still establishing standard processing and analysis methods, I knew that if I wanted to synthesize many published findings, I would need to re-process each study's raw data from scratch so the results would actually be comparable. I scoured the literature for any case-control (i.e. healthy vs. disease) study that reported publicly-available 16S gut microbiome data. I felt like a data detective at times, trying to find the data associated with a paper and figuring out what processing had been done to the data, which files belonged to which samples, and why sample sizes didn't match between my data and what was reported in the papers. I learned a lot in the process of downloading and processing these datasets: the magic of leaving myself thorough README's, the importance of just looking at the raw data and seeing if it makes sense, and the robustness of many 16S processing steps to my many small mistakes. I also learned to really appreciate well-documented and deposited data, and became more understanding of all of the moving parts required to report methods in a truly reproducible way.
Once I finally had the data downloaded and re-processed, it was time to start the re-analysis. Our idea was to just do the simplest possible analysis and and compare results across studies to find consistencies and patterns. For each study, I did univariate tests on the bacterial abundances between case and control patients. At first, I was actually pretty bummed because it didn't seem like much was that consistent: there weren't really any bacteria that were always associated with a given disease across all of its studies. And it was hard to know how many studies a bacteria needed to be significant in to be considered a "true" disease-specific association. Then one day, I was showing some colleagues the heatmaps of my p-values and complaining that I didn't really know how to interpret them given the between-study heterogeneity. In the plot, I had sorted the bacteria alphabetically by taxa (as a proxy for phylogeny), and the p-values looked pretty random. A colleague recommended that I sort the bacteria in a more "informative" way, for example by the overall effect direction of each bacteria across the studies. I made the changes to my code and plotted these new heatmaps for each disease separately, and the results were immediately obvious: my IBD p-value heatmap was almost all blue (health-associated) and the colorectal cancer heatmap was almost all red (disease-associated). Although the individual significant bacteria weren't necessarily consistent across studies of the same disease, there were clear patterns of associations that were!
In other words, we found that you can more precisely describe "dysbiosis" by instead describing certain consistent patterns of disease-associated microbiome shifts. These shifts differ in their directionality ("too many bad bugs" vs. "not enough good bugs"), magnitude (many significant associations vs. only a few), and specificity (always the same associated bacteria vs. different bacteria in different studies). We actually weren't able to quantify the specificity of shifts because we didn't have any disease with enough studies to make a strong statistical statement, but we think this will be an important idea to keep in mind as research moves forward. Describing disease-associated shifts with this framework is more useful than the general "dysbiosis" because it suggests possible approaches for microbiome-based interventions and therapeutics tailored to diseases with different types of shifts (we talk about this more in the paper).
Our second main question in the paper was whether there was some set of "core" health- or disease-associated microbes, which were significantly associated with health or disease no matter what disease was being studied. At first, I struggled to figure out how to define these non-specific responders: it wasn't clear how to deal with unobserved bacteria and how to make sure that the highly significant results (like many in the diarrhea studies) didn't overshadow all other patterns. In the spirit of our earlier analyses, however, we decided to do the simplest possible thing: simply count the number of diseases a given bacteria was significant in, and see which bacteria showed up in more than one disease. When we went back to each individual study and compared their individual results with this list of non-specific bacteria, we were taken aback to see that the majority of each study's associations overlapped with these non-specific responders. Basically, this means that in any given study, about half of the identified significant associations probably aren't specific to the disease of interest, and follow-up analyses or experiments will be needed to nail down which associations are truly disease-specific bacteria.

In addition to the scientific insights of this paper, my more personal takeaway from this project is in the power of combining data to help us do better science. In this work (and hopefully future case-control microbiome studies), contextualizing results across diseases helped us figure out which hypotheses were more likely to be disease-specific and maybe worth following up with for explorations of causality or diagnostic potential. Combining many studies also helped us come up with new hypotheses, such as the idea that there’s a shared response to health and disease, which you could perhaps use to develop smarter probiotics or antibiotics that could work across a variety of conditions. These insights wouldn’t have been possible without comparing across many datasets and diseases, which itself wouldn't have been possible without publicly available and well-documented data. I also gained a lot of appreciation for the work it takes to make data publicly available in a useful way. I learned that it's about a lot more than just uploading the data: you also have to make sure the data is connected to its metadata, that the metadata includes both clinical and technical information (i.e. both things like disease state as well as replicate numbers, data processing already performed, etc), and that samples are tracked throughout the entire processing steps. In fact, realizing how easy it is to forget to document very important steps in methods was the main motivation behind putting in the work to make all my analyses reproducible with a Makefile - I didn't trust myself to be thorough enough and so appreciated having a "make" command that would break if I'd missed any steps! That said, with all the current talk of reproducibility crises, I was actually really heartened to see that the majority of published findings were pretty reproducible, with only few discrepancies that you'd expect from different analysis methods.
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
I just found this fantastic blog post by the same author, Claire Duvallet. A must read IMO!
An empirical analysis of microbiome data availability
https://cduvallet.github.io/posts/2018/04/microbiome-data
By the way, congratulations on your 'Junior Research Parasite' award!
https://twitter.com/cduvallet/status/1082109581373964293