Building a predicative tool for synthetic gene networks
Published in Microbiology
Gene circuits lie at the center of synthetic biology for programming cellular functionality. To date, their design is largely based on experience and trial-and-error.
Written by Chen Liao and Ting Lu
Find the full story in Nature Microbiology: http://go.nature.com/2wMRttE
Synthetic biology is an interdisciplinary research field that utilizes engineered gene circuits to program novel cellular functionalities. Similar to electrical circuits used in numerous electronic devices, genetic circuits have been created to generate defined dynamics, rewire endogenous networks, sense environmental stimuli, and produce valuable molecules. Such a functional versatility of gene circuits offers enormous potential in advancing the basic understanding of biology as well as in empowering novel applications for practical purposes.
So far, most circuits are constructed through a trial-and-error manner, which relies heavily on a designer’s intuition and is often labor-intensive and inefficient. With the increase of circuit complexity, the lack of predictive design guidelines has become a major challenge in realizing the potential of synthetic biology.
Quantitative modeling of gene circuits serves as a natural choice to address the challenge, and has been extensively practiced. Currently, typical models regard gene circuits as isolated entities and focus only on the biochemical processes within the circuits. However, such a ‘standard’ paradigm is often incapable of quantitatively, or even qualitatively sometimes, describing circuit behaviors. It has received challenges from increasing experimental evidences suggesting that circuits and their host are intimately connected and their interactions can potentially impact circuit behaviors.
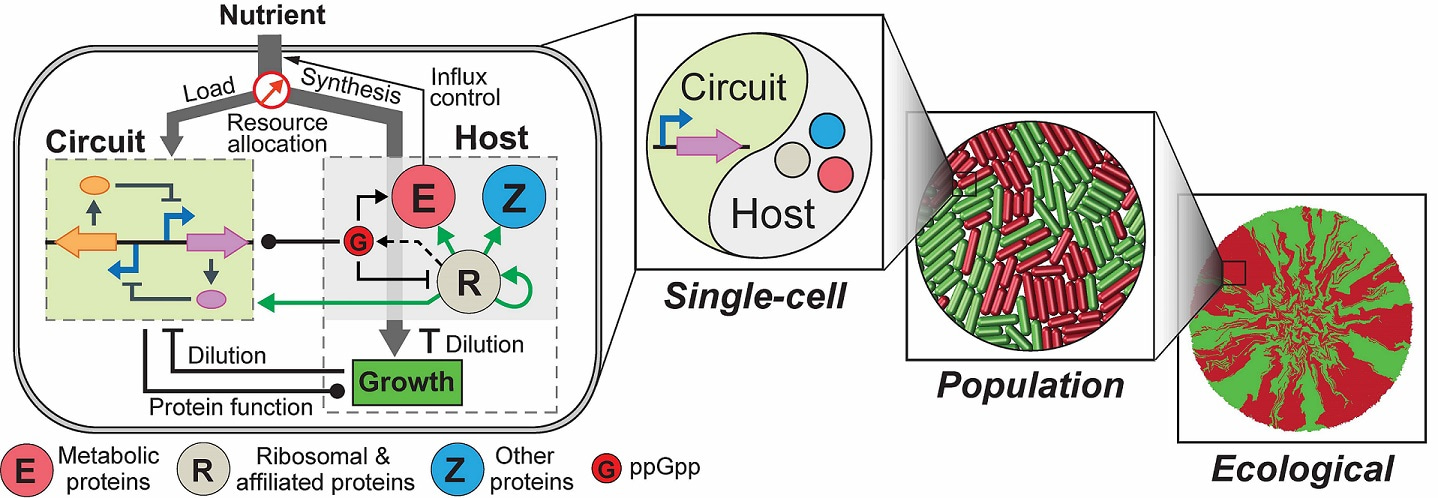
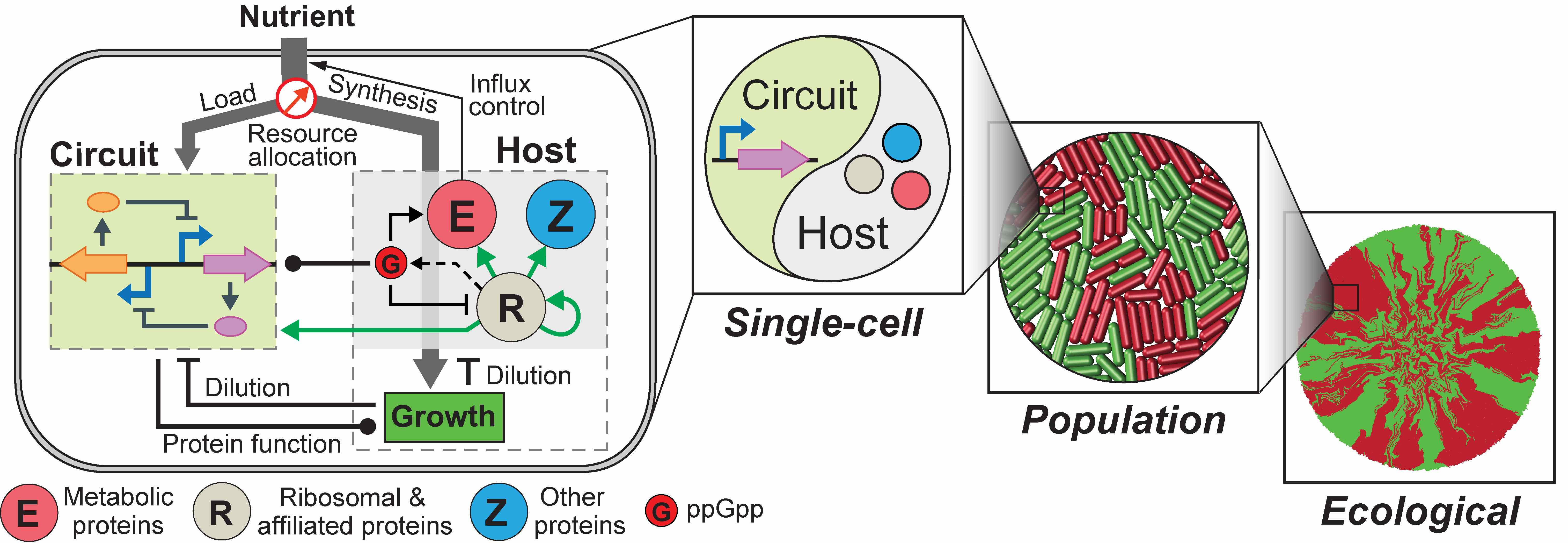
We recently addressed the challenge by constructing an integrated modelling framework for quantitatively describing and further predicting gene circuit behaviors by using Escherichia coli as a model host. The framework consists of a coarse-grained but mechanistic description of host physiology that involves dynamic resource partitioning, multilayered circuit-host coupling, and a detailed kinetic module of exogenous circuits.
We showed that, following training, the framework was able to capture and predict a large set of experimental data concerning the host and simple gene overexpression. For example, we found that ppGpp-mediated effects are the key to understand constitutive gene expression under environmental changes, including nutrient and antibiotic perturbations. To demonstrate its utility, we applied the framework to examine a growth-modulating feedback circuit whose dynamics is qualitatively altered by circuit-host couplings, and further revealed the behaviors of a toggle switch across scales from single-cell dynamics to population structure and to spatial ecology.
An important note to our framework is that, although the model was established using E. coli as the model host, it holds the encouraging promise to be generalized for describing multiple host organisms. For example, we found that, by varying only a single parameter several, the model successfully predicted key host metrics, including RNA-to-protein ratio, RNA contents per cell, and mean peptide elongation rate, for E. coli, Salmonella typhimurium and Streptomyces coelicolor.
This work advances our quantitative understanding of gene circuit behaviors, and benefits the transformation of gene network design from trial-and-error construction to forward engineering. By systematically illustrating key host cellular processes and multilayered circuit-host interactions, the work further sheds light on quantitative biology towards a better understanding of complex bacterial physiology.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in