Cancer predisposition signaling in Beckwith-Wiedemann Syndrome drives Wilms tumor development
Published in Cancer and Protocols & Methods
Background
Wilms tumor (WT), also known as nephroblastoma, is the most common pediatric kidney cancer. Children who have Beckwith-Wiedemann syndrome (BWS, OMIM 130650), a cancer predisposition syndrome, have an approximately 800-fold increased relative risk of developing WT. Therefore, tumor screening is recommended for patients with BWS by ultrasonography every three months through the 7th birthday. BWS is most often caused by improper epigenetic methylation on chromosome 11p15, and BWS is molecularly diagnosed by measuring differences in the methylation of this chromosomal region. The currently normal methylation range used to study 11p15 alterations is very broad, and at least two-thirds of WT have methylation alterations at chromosome 11p15, the BWS critical region. Due to these findings, our aim was to examine the molecular mechanisms underlying the link between BWS and WT oncogenesis based on methylation levels at chromosome 11p15.
Purpose
In this study, we present the first molecular multi-omics study of a cohort of patients with BWS and WT. The aim of this study is to identify the molecular signaling signatures in BWS driving WT oncogenesis. We also compared BWS tumor data from patients with BWS and WT (BWS-WT) to data from patients without BWS but with WT (nonBWS-WT) to identify putative drivers of WT in BWS kidneys. Within the nonBWS-WT group, there are some patients with normal methylation at chromosome 11p15 and some with altered methylation at chromosome 11p15. Thus, our study highlights the similarities and differences between altered-11p15 nonBWS-WT and BWS-WT tumors.
Methods: In this study, we used DNA methylation arrays, whole exome sequencing, and bulk RNA sequencing to elucidate the cellular signaling pathways involved in BWS-WT development. We compared our BWS cohort with nonBWS-WT, stratified by 11p15 status (altered/normal methylation at 11p15), to shed light on the genomic, methylomic, and transcriptomic changes that arise because of 11p15 alterations.
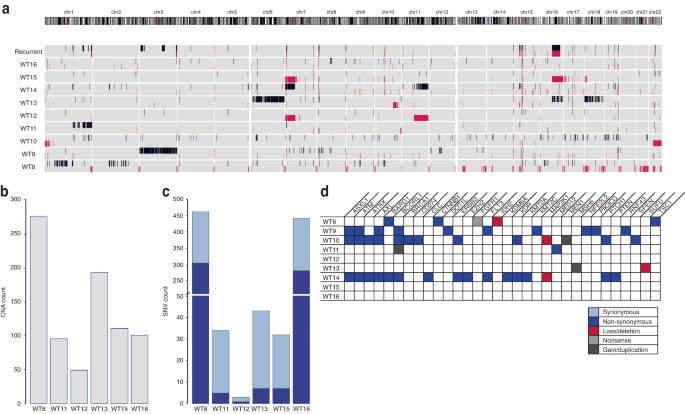
Findings
Recurrent mutations in genes associated with sporadic WT, such as TP53, CTNNB1, and WT1, were not observed in the BWS-WTs. However, we did find other single nucleotide variants (SNVs) in BCORL1, ASXL1, ATM and AXL genes in the BWS-WT. We defined a narrow methylation range to stratify nonBWS-WT into altered-11p15 nonBWS-WT and normal-11p15 nonBWS-WT. Once we divided the samples into these groups, global differential methylation analysis of BWS-WT showed enrichment of biological processes related to the Wnt signaling pathway, integrin signaling pathway, insulin receptor signaling pathway, and BMP signaling pathway. Examination of global differential methylation analysis of altered-11p15 nonBWS-WT showed a different profile, with enrichment of cell cycle processes, DNA damage checkpoint processes, TORC1 signaling, TOR signaling and Wnt signaling pathways. Expression analysis via RNA-sequencing revealed differential gene expression of Wnt signaling, along with other distinct pathways including: Notch signaling, BMP signaling, PPAR signaling, and NIK/NF-kappaB signaling pathways in BWS-WT compared to control kidneys. We also performed an interactome study with the enriched genes in BWS-WT, and we uncovered that CTNNB1 had the broadest range of interactions, and its overexpression was observed in BWS-WT. Interestingly, most of these upregulated pathways and interactions are reminiscent of an alteration in the stem/progenitor differentiation programming and nephron patterning, suggesting that BWS-WT may contain dysregulated nephron progenitor cells as a causal agent in disease development and/or progression. However, a dedicated model is required to explain the combinatorial role of these pathways in the earliest events of WT tumorigenesis.
Conclusion
While WT predisposition in BWS is well-established, as are 11p15 methylation alterations in nonBWS-WT, this study is the first to focus on stratifying tumor genomics by 11p15 methylation and BWS status. We have identified pathways unique to BWS-WT and altered-11p15 nonBWS-WT that are differentially regulated by altered methylation gene expression. This observation indicates that altered-11p15 nonBWS-WT and BWS-WT have some common features, but they also have unique gene signatures attributed to the epigenetic predisposition in BWS. WT oncogenesis can be seen as a spectrum of molecular signaling signatures and differential tumor mutational burdens (Figure 1) seen in BWS and nonBWS cohorts. This is the first study that highlights these features of WT oncogenesis based on 11p15 alterations. Further investigation of our findings may serve to identify novel diagnostic or therapeutic targets to combat WT oncogenesis.
.png "Spectrum of molecular signaling pathways in WT oncogenesis.")
Follow the Topic
-
British Journal of Cancer
This journal is devoted to publishing cutting edge discovery, translational and clinical cancer research across the broad spectrum of oncology.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in