Carbon-Carbon Coupling on a Metal Non-metal Catalytic Pair
Published in Chemistry


The use of electrochemical CO2 reduction reaction (CO2RR) to synthesize carbon-based fuels in conjunction with renewable electricity represents a promising strategy to address current concerns regarding global energy shortages and environmental issues such as climate change 1. It can provide ways to integrate carbon capture, utilization, and storage in energy cycles while maintaining controllable levels of atmospheric CO2. Liquid compounds are more preferred CO2RR products because of their high energy densities (e.g., -1366.8 kJ/mol for CH3CH2OH) and ease of storage when serving as substitutes for gasoline.2 To date, the majority of catalysts found able to generate Cn (n》2) liquid products from the CO2RR with appreciable reaction rates are based on Cu 3. Unfortunately, due to a lack of comprehensive understanding of C-C coupling process over Cu-based catalysts, the control of the C-C bond formation is challenging a4, 5, which has greatly limited the development of highly selective electrocatalyst to reduce CO2 to Cn (n》2) liquid products.
In this study, Prof. Bin Liu’s group at City University of Hong Kong proposes a Cu-free, Sn-based tandem electrocatalyst composing of SnS2 nanosheets and single Sn atoms coordinated with three oxygen atoms on three-dimensional (3D) carbon (Sn1-O3G), in which the SnS2 nanosheets produce formate intermediates and the Sn1-O3G sites generate *OCO(OH) bicarbonate intermediates, followed by C-C coupling over the dual active centers comprising Sn and O atoms (Fig. 1). Based on this strategy, an ethanol selectivity of more than 70% was achieved over a wide potential range from -0.6 to -1.1 VRHE. This catalyst was found to maintain 97% of its initial activity after 100 hours of continuous reaction at a geometric current density as high as 17.8 mA·cm−2. The C-C bond formation mechanism on this developed tandem catalyst was elucidated by combining isotope (13CO2 and H13COOH) labelling experiments with extensive density functional theory (DFT) studies, providing a highly efficient C-C coupling pathway for ethanol synthesis via the CO2RR. It is anticipated that this work will lead to new applications for atomically dispersed catalysts with dual active centers in multi-step chemical reactions.
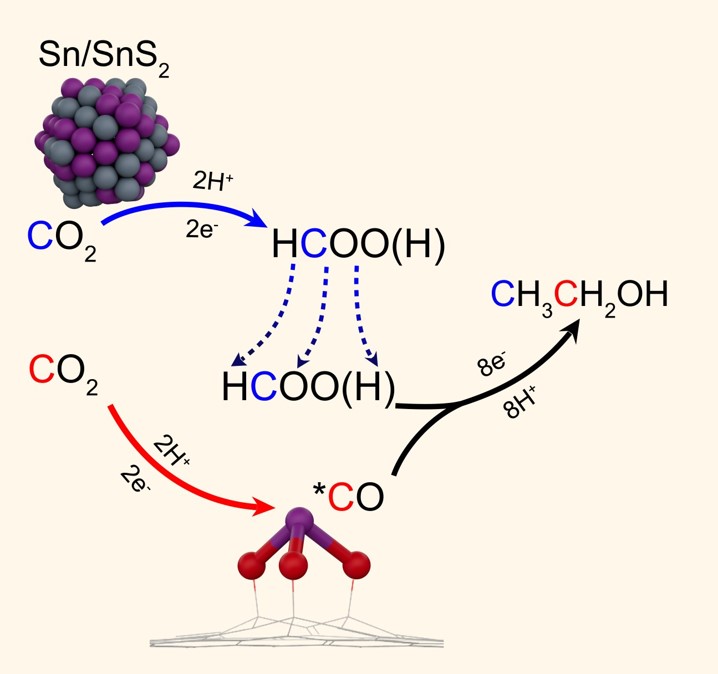 Fig. 1 The CO2RR to CH3CH2OH pathway on SnS2/Sn1-O3G.
Fig. 1 The CO2RR to CH3CH2OH pathway on SnS2/Sn1-O3G.
The electrochemical CO2RR performance was firstly evaluated using chronoamperometry in a H-type cell filled with CO2-saturated 0.5 M KHCO3 solution. The SnS2/Sn1-O3G catalyst was stable during CO2 reduction to ethanol, maintaining approximately 80% ethanol selectivity at -0.9 VRHE and 97% of the initial activity after 100 hours of continuous reaction (Fig. 2a). It is worth noting that no other C2 products (such as ethylene or acetate) were detectable over the entire potential range applied during the CO2RR. Compared to state-of-the-art catalysts for electrochemical CO2 reduction to ethanol (Fig. 2b), the Sn-based tandem catalyst shows outstanding CO2-to-ethanol performance.
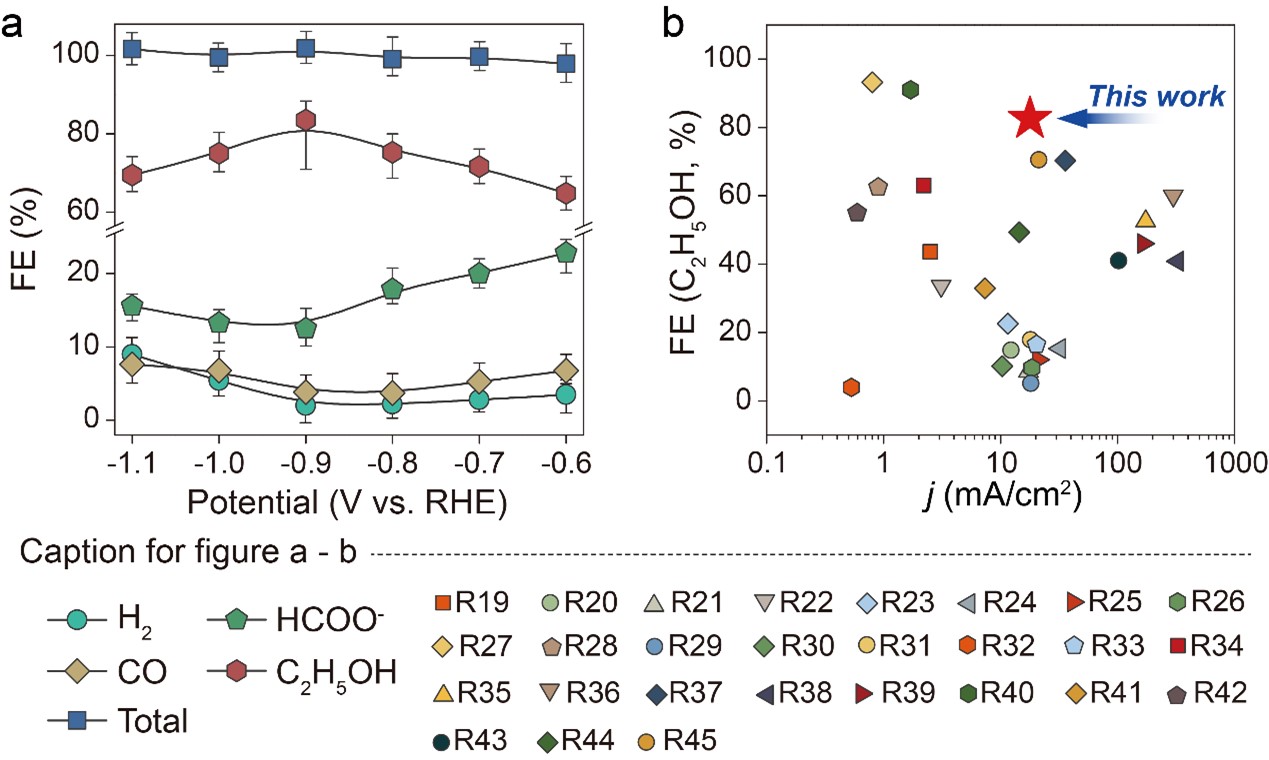 Fig. 2 (a) FE values with error bars indicated (the FE was measured in 2 h interval at each potential). (b) Comparison of the CO2RR performance to ethanol in aqueous system.
Fig. 2 (a) FE values with error bars indicated (the FE was measured in 2 h interval at each potential). (b) Comparison of the CO2RR performance to ethanol in aqueous system.
A series of control experiments, including comparison of different catalysts and isotope labeling experiments (Fig. 3), were performed to understand the role of each component in SnS2/Sn1-O3G for the CO2RR. These results demonstrate the importance of HCOOH in C-C coupling to form ethanol on O atoms coordinated Sn catalytic sites (as shown in Fig. 1).
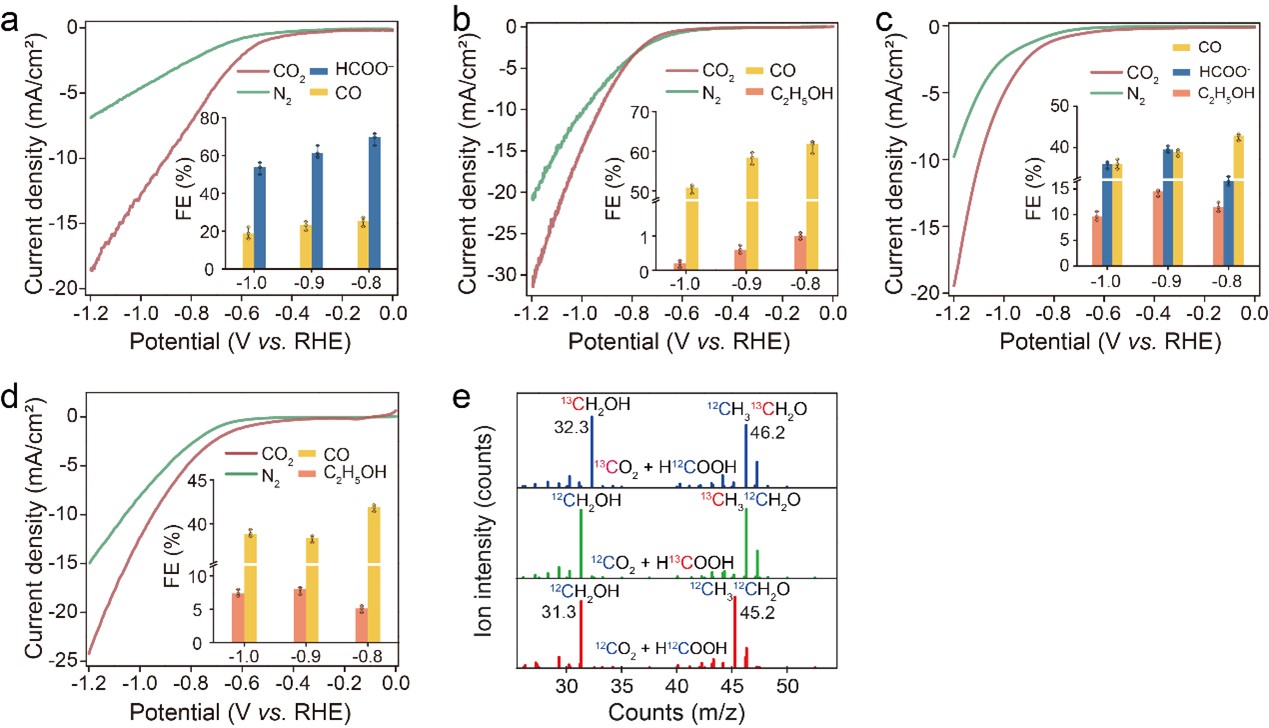 Fig. 3 (a) and (b) LSV curves of SnS2 nanosheets and Sn1-O3G. Inset displays the FE. (c) LSV curves and FE data for a physical mixture of SnS2 and Sn1-O3G with a 1:1 mass ratio. (d) LSV curves and FE data for Sn1-O3G with HCOOH addition. (e) Mass spectra of the CO2 reduction products using 13CO2 (top) or H13COOH (middle) as the reactant, biased at -0.9 V vs. RHE, or using 12CO2 and H12COOH (bottom) as the reactants under the same electrochemical conditions. The error bars indicate the between the maximum and minimum values of three different tests.
Fig. 3 (a) and (b) LSV curves of SnS2 nanosheets and Sn1-O3G. Inset displays the FE. (c) LSV curves and FE data for a physical mixture of SnS2 and Sn1-O3G with a 1:1 mass ratio. (d) LSV curves and FE data for Sn1-O3G with HCOOH addition. (e) Mass spectra of the CO2 reduction products using 13CO2 (top) or H13COOH (middle) as the reactant, biased at -0.9 V vs. RHE, or using 12CO2 and H12COOH (bottom) as the reactants under the same electrochemical conditions. The error bars indicate the between the maximum and minimum values of three different tests.
Furthermore, DFT calculations were performed to map the CO2RR pathway during the formation of ethanol. The results show that, during the CO2 reduction process, the protonation of Sn1-O3G-OH pre-catalytic sites is exergonic (Fig. 4). A differential charge distribution analysis of 3 indicates that the electron density is higher on the Sn atom, such that the formation of a bond with HCOOH is favored. After formation of this preferred intermediate, HCOOH is adsorbed at the Sn site via the C atom, and the carbonyl group in HCOOH* is hydrogenated and dehydrated to generate intermediate 4, with a free energy barrier of 0.52 eV. Following the drastic reduction of all carbonyl groups in 4, intermediate 5 is formed as the precursor for C-C coupling. The free energy barrier to C-C bond formation is 0.13 eV, which is much lower than those of Cu-based catalysts (> 1.0 eV). Thus, intermediate 6, which contains a Sn-C-C-O quaternary ring, is obtained. As reduction proceeds, with exergonicity of 0.99 eV, the Sn-C bond breaks, then forms the *CH3CHOH intermediate (7), followed by O-C bond cleavage, and finally releases CH3CH2OH from the active site. Owing to the flexibility of the bond between the active O atom and the C on the support, the Sn1-O3G is distinguished by the dual active sites of Sn and O that are able to adsorb different C-based intermediates, providing a novel platform for C-C bond formation during the CO2RR to produce ethanol.
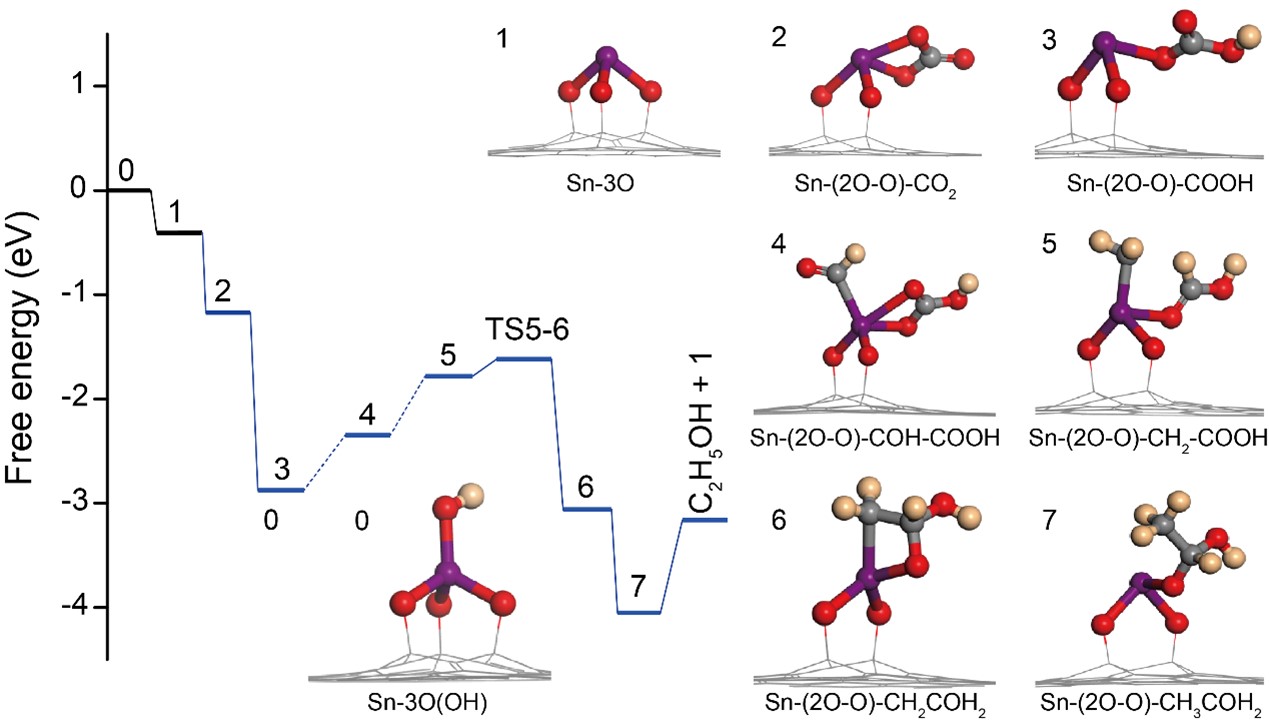 Fig. 4 Reaction energy profiles and the corresponding intermediate structures (0 to 7) for the formation of ethanol via the CO2RR on the Sn1-O3G catalyst.
Fig. 4 Reaction energy profiles and the corresponding intermediate structures (0 to 7) for the formation of ethanol via the CO2RR on the Sn1-O3G catalyst.
In this work, Dr. Jie Ding, Prof. Yanqiang Huang, Prof. Bin Liu and co-authors overcome the restrictions of Cu-based catalysts, achieving highly selective C-C coupling on a metal non-metal catalytic pair. The search for more efficient catalysts with dual-functional catalytic sites should be pursued through high-throughput experiments and theoretical calculations. In-depth study on factors that affect the residence time of intermediates, such as the pore structure of the support for the dual functional Sn1-O3G sites, will deepen the understanding of the C-C coupling process. Last but not least, cascade catalysis based on the concept of catalytic pairs is expected to be extendable in C-X (X = N or S) coupling to prepare new chemicals, such as urea and alanine.
References
- Mehling, M. A., Metcalf, G. E. & Stavins R. N. Linking Climate Policies to Advance Global Mitigation. Science 359, 997 (2018).
- Li, C. W., Ciston, J. & Kanan, M. W. Electroreduction of Carbon Monoxide to Liquid Fuel on Oxide-Derived Nanocrystalline Copper. Nature 508, 504 (2014).
- Garza, A. J., Bell, A. T. & Head-Gordon, M. Mechanism of CO2 reduction at copper surfaces: pathways to C2. ACS Catal. 8, 1490 (2018).
- Zhuang, T.-T. et al. Steering post-C-C coupling selectivity enables high efficiency electroreduction of carbon dioxide to multi-carbon alcohols. Nat. Catal. 1, 421 (2018).
- Nitopi S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610 (2019).
Follow the Topic
-
Nature Energy
Publishing monthly, this journal is dedicated to exploring all aspects of this on-going discussion, from the generation and storage of energy, to its distribution and management, the needs and demands of the different actors, and the impacts that energy technologies and policies have on societies.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in