Combining Methods of Gene Editing for Applications in Biotechnology
Published in Bioengineering & Biotechnology

In 2012, I came across the concept of bacterial expressions systems within a practical setting, when Escherichia coli (E.coli) organisms were used to produce recombinant human tropoelastin, at the Department of Biochemistry, University of Sydney 1. The synthetic protein tropoelastin is a precursor to the protein elastin, naturally present in the extracellular matrix (ECM)2 subsequently used for bioengineering applications with biomaterials3,4. Recombineering is a genome engineering method primarily used within living E.coli bacteria, allowing easy manipulation of bacterial chromosome for metabolic engineering and production of a wide range of biochemicals 5. Integration of systems biology, synthetic biology and evolutionary engineering has broadly enabled an assortment of genetic tools and protocols for efficient, cost-effective manipulation of E.coli in research and industrial applications6. To execute recombineering protocols, a basic knowledge of molecular and microbial techniques is required. This is a summary on current advancements of gene editing and its applications in synthetic biology.
New era of synthetic biology
At present, rapidly evolving gene technologies are anticipated to play a significant role towards addressing growing global challenges. Genetic engineering has revolutionized studies in molecular biology for the last 30 years since the discovery of restriction enzymes 7. Restriction enzymes are an important tool in genomic research, akin to molecular scissors that cut DNA at a specific site and create a space to insert foreign DNA for editing purposes. Recombinant DNA technology, introduced in the 1970s created a pathway to insert foreign genes into E.coli host cells via shuttle vectors (a molecular vehicle) called plasmids, for gene expression and recombinant protein production (Figure 1). Owing to intrinsic limitations of plasmid-based applications including low yield of proteins, deleterious dosing of genes and significant metabolic burden to host cells, foreign genes were later integrated directly into the E.coli genome instead. Thus over the past decade, many advancements were made in gene engineering; starting with the replacement of in vitro (in-lab) recombinant genetic engineering technology with in vivo (in organism) recombineering technology, for more precise, rapid and practically efficient results 8. Importantly, the in vivo recombineering technology does not require restriction enzymes or DNA ligases, which were an essential component of classical genetic engineering9.
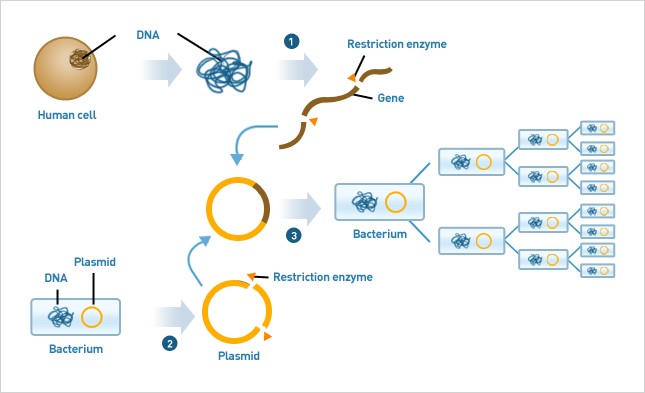
Figure 1:.Using restriction enzymes to cut a DNA fragment of interest and paste it with ligases into the plasmid "vectors". The pioneering work introduced in the early 1970s is still a widely used approach (Figure from AbFrontier).
Recombineering
Since its inception in 1998, recombineering or recombinogenic engineering has allowed direct engineering of bacterial chromosome within the organism (in vivo) by homologous recombination9,10. Homologous recombination is the process by which segments of DNA are exchanged between two DNA molecules through regions of identical sequence, to result in the new combination of genetic material7. Bacteria such as E.coli, encode their own homologous recombination systems, while viruses (or phages) that inhabit bacteria carry their own recombination functions and these two systems can work with or independently of each other11. Biotechnology has taken advantage of such phage proteins within bacteria to make use of homologous recombination for DNA engineering in E.coli, thus enabling a chosen DNA region to be cloned from a complex mixture, without restriction enzymes or prior isolation 11,12. The most popular phage systems (virus systems inhabiting bacteria) are known as RecET based on genes encoded by RAC prophage and lambda Red, based on the homologous recombination system of bacteriophage λ (lambda), present in some E.coli strains 13.
The segments of donor DNA used for recombineering can either be linear double stranded DNA (dsDNA) or synthetic single stranded oligonucleotides (ssDNA oligos)13, efficiently used in a variety of approaches (to create single-base changes, small deletions, large deletions and small insertions) and engineer DNA in E.coli9. The linear donor DNA substrate with desired change can be introduced by electroporation into bacterial strains to express the recombination functions, allowed to recover for a period of time and screened to identify recombinant colonies5,8. The phage-based recombination system can catalyze homologous recombination between linear DNA and a replicon such as phage λ, bacterial chromosomes, plasmids and bacterial artificial chromosomes (BACS)9. Researchers strategically plan the recombineering pathway most appropriate to produce desired construct based on several different, extensive protocols in molecular biology8. Compared to classical genetic engineering, the editing process between a linear donor DNA substrate and replicon (BAC) is comparatively less labour-intensive and more efficient via recombineering (Figure 2).
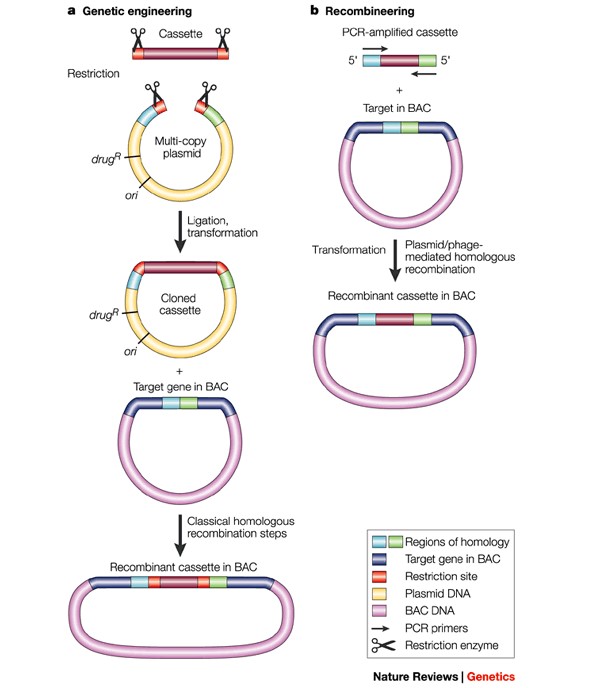
Figure 2: Modifying the BAC clones in E.coli. a) In traditional genetic engineering, DNA fragments are cut and rejoined using restriction enzymes and DNA ligase, for classical homologous recombination, b) problems associated with the traditional approach have been overcome by the use of phage-mediated recombination in recombineering technology (Figure from reference12).
Lambda Red Recombineering in Escherichia coli
Linear DNA introduced into bacterial systems are normally prone to degradation by bacterial nuclease enzymes7. Over the past decade, lambda Red recombineering has been used as a powerful technique for precisely defined chromosomal alterations in E.coli10. Three phage-derived lambda Red proteins: Gam, Exo and Beta are necessary to complete dsDNA recombination7,10; Gam prevents degradation of foreign linear double stranded DNA by the E.coli nucleases, Exo degrades dsDNA to form a single stranded DNA (ssDNA) and Beta binding facilitates recombination (Figure 3). The exact mechanism on how a desired construct recombines with the chromosome in the presence of the three lambda Red proteins has been highly debated 14. Furthermore, λ Red DNA-based engineering is the only method that allows multiplexing (sequential editing) in one step by direct electroporation, thereby used extensively as an editing tool for Multiplex Automated Genome Engineering (MAGE)13.
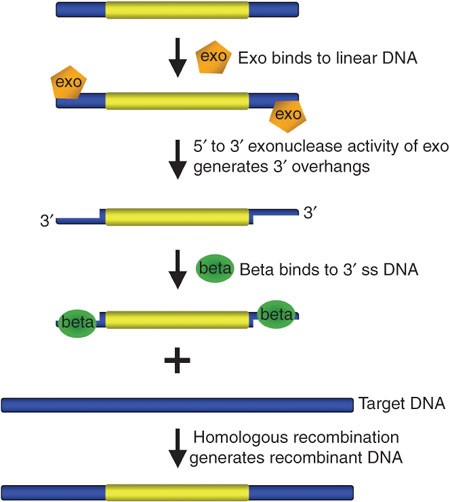
Figure 3: Overview of bacteriophage λ red recombination system used for recombineering. 1) Exo protein has exonuclease activity (5' to 3') generating 3' overhangs on linear DNA. 2) Beta binds the single-stranded DNA (3' overhangs), promotes ss-annealing and generates recombinant DNA. 3) An additional protein Gam (not shown here) prevents the E.coli enzymes from degrading double-stranded linear DNA fragments required for dsDNA recombineering (Figure from reference5).
Coupling CRISPR/Cas9 system with Lambda Red Recombineering in E.coli
Despite novel techniques incorporated, the gene and protein yield with such bacterial expression systems remain small/insignificant leading to a bottleneck in metabolic engineering and systems biology. For instance, efficiency of short ssDNA/dsDNA oligonucleotide-mediated recombineering is highest for short genome modifications, while larger modifications occur with significantly lower frequency (<1%)15, necessitating screening via polymerase chain reaction (PCR; an oligonucleotide amplification technique in molecular biology) to identify for the desired mutations that do not result in a clear phenotypic change15. Furthermore, to select for recombinant clones, recombineering methods rely on insertion of antibiotic markers to the gene, which must be removed prior to further modifications, thereby causing scars in the genome. Scars promote genome rearrangement after several rounds of modifications making it difficult to further engineer the strain. These limitations can be overcome by coupling lambda red recombineering with the currently powerful tool in molecular biology; clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system, which has proven ability to edit genes of a variety of organisms including Eukaryotes, Prokaryotes, higher plants and even human cell lines13,15. By function, Cas 9 is a programmable nuclease that can mediate a blunt double-stranded break (DSB) at almost any target DNA loci missing the desired mutation and since DSB is lethal, this will result in overall cell death thereby acting as a better selection/screening procedure for cells possessing intended modification generated via homologous recombination vs the wild-type (non-mutant cells)13. Since CRISPR/Cas9-coupled recombineering doesn’t therefore rely on antibiotic marker for selection, these gene replacement mutants are ideally “scar”-less15. The process is also performed in a shorter period of time than previous protocols.
In 2016, the technology was advanced one step further, to integrate automation (MAGE) for a combined; CRISPR/Cas9 and λ Red recombineering-based MAGE technology, known as CRMAGE15. This system can be multiplexed to enable introduction of at least two mutations in a single round of recombineering with similar efficiencies, for multiple engineering rounds per day15. In a single round of CRMAGE recombineering, scientists were able to achieve close to 98% efficiency for a single point mutation which is unprecedented compared to previous efficiency of 5% using traditional MAGE. The study further demonstrated that it’s possible to modulate protein translation with relatively high frequency (from 6% to 70%), within the cell population as well15. Any regulatory element can thereby be inserted to modulate either gene expression or protein translation. Technically, it is even possible to obtain a clean, final recombinant strain by removing the entire CRMAGE editing system from the bacterial population after recombineering15. To speed up the design process for editing, the research team have facilitated a web-based tool that can predict the oligonucleotides for use in the editing process. The CRMAGE technique will enable generation of multiple mutations in a single cycle and multiple cycles within a single working day, to significantly increase the daily capacity of engineering the genome of E.coli and other bacterial strains, for potential high-throughput, research and industrial applications in biotechnology.
Poster Image: Screenshot from - 'What is genetic modification?' animated video of The Royal Society, available via YouTube.
References:
- Martin, S. L., Vrhovski, B. & Weiss, A. S. Total synthesis and expression in Escherichia coli of a gene encoding human tropoelastin. Gene 154, 159-166 (1995).
- Vrhovski, B. & Weiss, A. S. Biochemistry of tropoelastin. Eur J Biochem 258, 1-18 (1998).
- Waterhouse, A. et al. The immobilization of recombinant human tropoelastin on metals using a plasma-activated coating to improve the biocompatibility of coronary stents. Biomaterials 31, 8332-8340 (2010).
- Jeewandara, T. M. Bioengineering cobalt chromium cardiovascular stent biomaterial for biofunctionalization. Cold Spring Harbor Laboratory Press. bioRxiv Preprint, 1-17 (2016).
- Sharan, S. K., Thomason, L. C., Kuznetsov, S. G. & Court, D. L. Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc 4, 206-223, (2009).
- Chen, X. et al. Metabolic engineering of Escherichia coli: a sustainable industrial platform for bio-based chemical production. Biotechnol Adv 31, 1200-1223, (2013).
- Sawitzke, J. A. et al. in Methods in Enzymology 421, 171-199 (2007).
- Thomason, L. C., Sawitzke, J. A., Li, X., Costantino, N. & Court, D. L. in Current Protocols in Molecular Biology (John Wiley & Sons, Inc., 2001).
- Zhang, Y., Buchholz, F., Muyrers, J. P. P. & Stewart, A. F. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet 20, 123-128 (1998)
- Murphy, K.C. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol 180, 2063-2071 (1998).
- Zhang, Y., Muyrers, J. P. P., Testa, G. & Stewart, A. F. DNA cloning by homologous recombination in Escherichia coli. Nat Biotech 18, 1314-1317 (2000).
- Smith, C. Cloning and mutagenesis: tinkering with the order of things. Nat Meth 4, 455-461 (2007).
- Pyne, M. E., Moo-Young, M., Chung, D. A. & Chou, C. P. Coupling the CRISPR/Cas9 System with Lambda Red Recombineering Enables Simplified Chromosomal Gene Replacement in Escherichia coli. Appl Environ Microbiol 81, 5103-5114, (2015).
- Mosberg, J. A., Lajoie, M. J. & Church, G. M. Lambda Red Recombineering in Escherichia coli Occurs Through a Fully Single-Stranded Intermediate. Genetics 186, 791-799, (2010).
- Ronda, C., Pedersen, L. E., Sommer, M. O. A. & Nielsen, A. T. CRMAGE: CRISPR Optimized MAGE Recombineering. nature Scientific Reports 6, 1-11 (2016)
I am an interdisciplinary researcher with a strong commitment to bioengineering, biochemistry, organ-chips and molecular biology. As well as biomechanics and biomineralization in the broader context of medicine. I completed my PhD at the University of Sydney Australia in December 2016, and travel often, find me on Twitter and irl.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in