Dickkopf-1 (DKK1) drives growth and metastases in castration-resistant prostate cancer
Published in Cancer
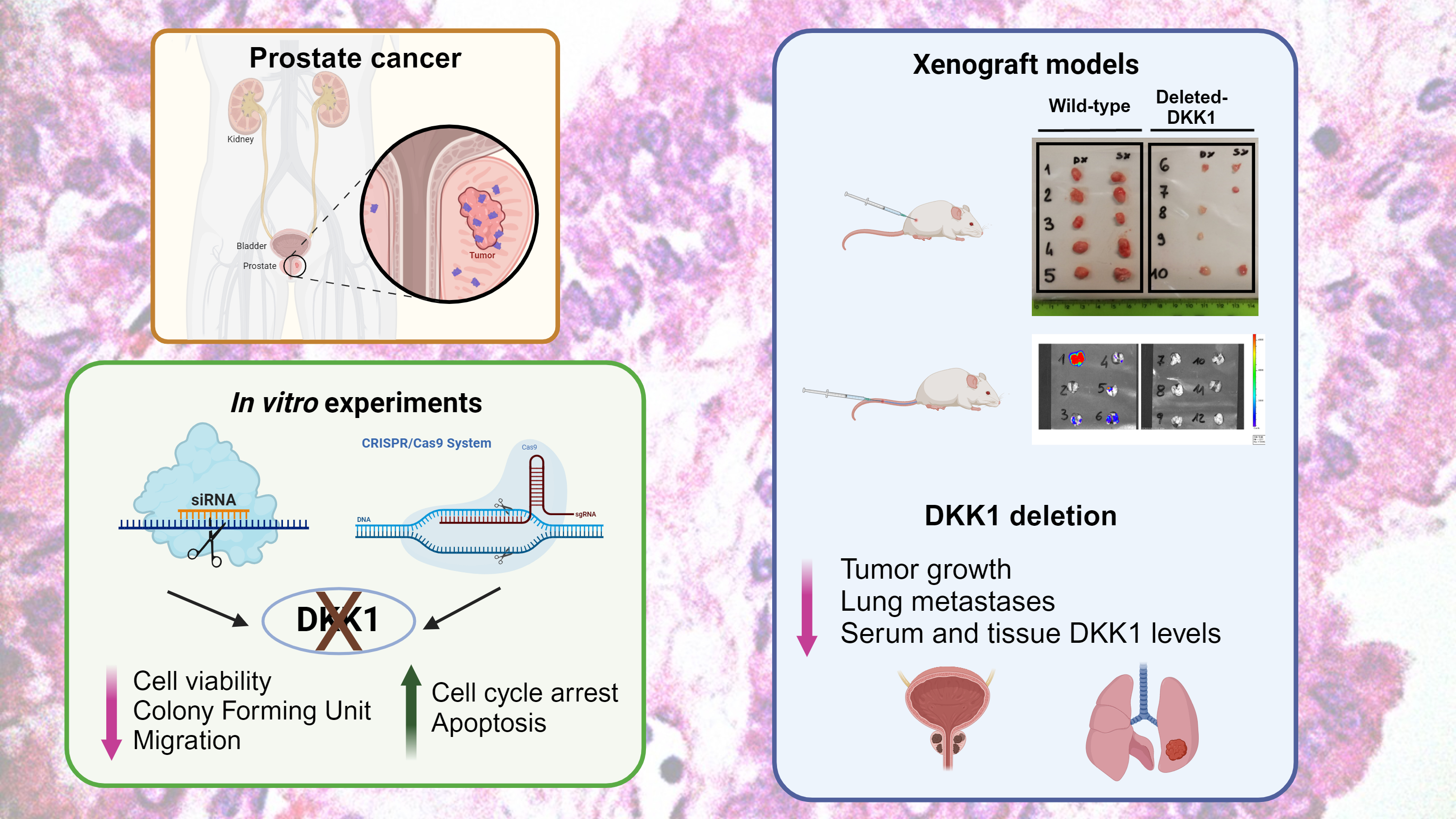
Prostate cancer is the second most frequent type of cancer in men worldwide and the fifth cause of cancer mortality. When the disease is confined to the prostate, it is well controlled by surgery and radiotherapy; but unfortunately, advanced or metastatic tumors have a poor prognosis and remain a fatal disease, despite the existence of multiple treatments. Therefore, a better understanding of the biology if prostate cancer and the identification of circulating molecular markers that could early predict progression, it’s advising and it could change the natural history of the disease. DKK1 is a secreted protein that controls the Wnt signaling pathway, involved in proliferation, stem cell maintenance, tissue commitment and differentiation. DKK1 has been showed to be highly expressed in a wide variety of cancers, suggesting that it may be involved in tumorigenesis and metastases as well.
Key findings
In prostate cancer the role of DKK1 is still debated; based on this, the aim of our work was to investigate the role of DKK1 in growth and migration of prostate cancer cells expressing high levels of DKK1, in in vitro and in vivo models.
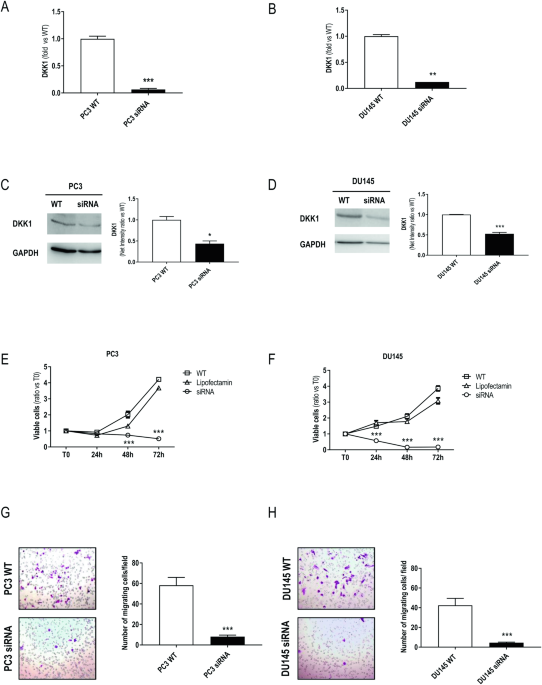
To exploit the role of DKK1 in driving tumor progression and metastatization, we first silenced DKK1 in two different prostate cancer cell lines. We used specific siRNAs, small oligonucleotides that specifically bind DKK1 mRNA, determining its degradation. In DKK1 silenced cells we observed growth arrest and a significant reduction of migration.
To further confirm our observation, we generated cells with stable DKK1 deletion by CRISPR/Cas9 editing technology. This system employs a Cas enzyme for cutting the target sequence and a single guide RNA, which binds to the target sequence. DKK1 deletion, as observed in silenced cells, determined a significant reduction of cell growth, and inhibition of cell migration. Moreover, DKK1 deletion resulted in the modulations of some genes involved in tumor metastases.
Finally, we created a xenograft mouse model using wild-type and DKK1-deleted cells. DKK1-deleted tumors exhibited slower growth, smaller volume and reduced weight than tumors generated by wild-type cells. In addition, different genes involved in metastases formation, were down-regulated in tumors generated with DKK1-deleted cells. Wild-type cells were able to promote formation of lung metastases, being clearly evident in lungs at sacrifice; on the contrary, only one mouse inoculated with DKK1-deleted cells developed metastases. Interestingly, in mice inoculated with wild-type cells, serum DKK1 levels were significantly higher than in mice inoculated with DKK1-deleted cells.
Conclusions
In this study, we provide evidence of a key role of DKK1 in prostate tumor growth and metastases formation, in vitro and in vivo. Our study contributes to a better knowledge of the molecular mechanisms underlying progression of prostate cancer, by reinforcing the previous evidence of DKK1 role in tumorigenesis. Specifically, we have shown the critical role of DKK1 in the tumor growth and metastasis formation in prostate cancer, confirming the possible role of DKK1 as circulating marker in prostate cancer and offering the preclinical rationale for new therapeutic strategies that target this gene.
Follow the Topic
-
Cancer Gene Therapy
The essential gene and cellular therapy resource for cancer researchers and clinicians, keeping readers up to date with the latest developments in gene and cellular therapies for cancer.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in