Dissecting gene conversion by dissecting tetrads
Published in Ecology & Evolution

The paper in Nature Ecology & Evolution ishere: http://go.nature.com/2zou7f2
This paper is one in a series between long term collaborators: one partner is an experimental genomics lab in China (Sihai Yang and Dacheng Tian as lab head), the other an evolutionary bioinformatics group in the UK (led by Laurence Hurst). For this paper, we were joined by colleagues in Nice, France who contributed invaluable resources and knowledge. The China-UK collaboration is focused on understanding the causes of genetic variation, e.g. mutation and recombination. We have studied by parent-offspring sequencing the causes of mutation rate and recombination rate variation in several taxa (honey bees, bumble bees, Arabidopsis, rice and peach, each chosen to test particular hypotheses). In this paper, we were interested to better understand the role and evolutionary importance of gene conversion.
We have been intrigued by gene conversion for a few years now. When we were studying the recombination landscape of honey bees, we noticed that they have very low non-crossover (NCO) rates compared with the very high crossover (CO) rates. We thought at the time that this might mean that organisms have to choose between crossing over or non-crossover but they cannot increase both. Of course, it could be that the marker density (~0.3% in bees) is low so that some NCO events couldn’t be detected, so bees aren’t the best to study this problem. The present paper suggests we were wrong about CO and NCO trading off and that the two positively correlate across taxa: species with a high CO rate tend to have a high NCO rate.
A further motivation came from contradictory results in other species. Studies in Arabidopsis have given some mixed signals regarding the rate of gene conversion and similarly in yeast different studies come to different conclusions about the extent to which gene conversion might be biased (a GC<->AT mismatch in a conversion tract could be repaired in a biased or unbiased fashion). To some degree these disagreements might be owing to subtle method issues. One of the few really solid analyses of gene conversion is from a previous study in yeast by Steinmetz’s group. But is yeast a representative species as regards meiotic gene conversions? Indeed, with the best next-generation sequencing technology can we replicate the results of this seminal study?
Knowing this challenge and opportunity, we set to study meiotic gene conversions, hoping to reveal their landscape with the aid of high throughput genome sequencing. The first step was to select the best materials for the job. In no small part this means picking the right organism – or rather, organisms. As we are interested in the comparative genomics of gene conversion we thought it would be better to consider several species. We don’t think any group has attempted this before, but it is now possible with the latest sequencing technology. Our idea was to provide a study in which all taxa were treated using the same method, so any differences would be unlikely to be owing to method differences.
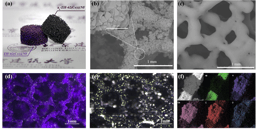
In our prior analyses, we have picked organisms to test particular hypotheses (e.g. to look for recombination induced mutation we compared highly recombinogenic honey bees with their lesser recombining relative the bumble bees, to look for an association of recombination with domestication we compared wild and domesticated peaches etc.). In this analysis the choice was much more determined by convenience. The organisms would need to have a good reference genome. In addition, ideally, (i) the organisms should be haploid, so that the sequencing is more accurate and analysis is easier; (ii) the four products of a given meiosis can be physically separated and sequenced individually, so that we can detect both NCO gene conversion and CO associated gene conversion (tetrad analysis is a gold standard for gene conversion), (iii) and finally, a high diversity cross is needed to provide enough markers to detect gene conversion (prior evidence suggests that the tracts could be really quite short – possibly just tens of base pairs - so the higher the marker density the better). We selected four organisms, the budding yeast Saccharomyces cerevisiae, the multicellular fungus Neurospora crassa, the unicellular plant Chlamydomonas reinhardtii, and the multicellular plant Arabidopsis thaliana (Figure below). Yes, Arabidopsis isn’t haploid but you can still do tetrad analysis. These four were tractable and diverse. They also allowed us to generate new data and to provide us with a sanity check by trying to replicate prior results.
The crossing and tetrad dissection in these organisms isn’t easy. Indeed, our lab has never used green algae nor Neurospora. As we don’t have any equipment to separate single cell spores, all the separation was done in the traditional way by hand picking with a glass needle under a regular microscope. We have, however, talented and persistent students to help with the experiments. After countless failed attempts, we eventually found the knack and got the spores separated.
With the single spores dissected we could grow these up and perform large scale whole genome sequencing. Crunching the data (which needs some care), we could estimate rates of both CO gene conversion and of NCO gene conversion. As a side product, we discovered that Neurospora appears to have motifs enriched at recombination breaks points (you can find this buried in the supplementary information) and could provide a recombinational map for all the species. We found three orders of magnitude difference in the percentage of markers converted per meiosis. We failed to find a universal GC-bias in the direction of gene conversion (or at least not that we could detect). In this regard, our study couldn’t replicate the very weak AT->GC conversion bias that Steinmetz’s group found in yeast, although we found very similar rates of gene conversion. If anything, our data suggests the opposite bias. We still don’t understand why the studies differ.
More generally, our study seems to leave more questions than answers. Why does the rate of gene conversion vary so drastically between species? Part of the difference between species follows de facto from differences in recombination rate, but tract lengths also show an order of magnitude variation. In yeast, that has the highest recombination rate, we also see the longest tracts – it is almost as if they are trying to maximize the amount of gene conversion. Does meiotic gene conversion play some more important roles in evolution that we didn’t know of? We liked Bengtsson’s idea that, if conversion is AT->GC biased, gene conversion is there to correct a universal GC->AT mutation bias (that we and others repeatedly find). While this model might apply in mammals and birds, except for NCO events in Chlamydomonas, we found no good evidence for it in our species. But why just NCO events and why just in Chlamydomonas? Bengtsson as it happens is a good friend of the UK wing of this collaboration, which is how we knew about his very interesting, but largely overlooked, hypothesis. It would have been a very elegant explanation for recombination were it true.
The apparent lack of GC bias in conversion in our species is puzzling. We have assumed for quite a while that conversion is GC biased and so this explains why GC content is highest in the most recombinogenic parts of genomes. This model works well for mammals and birds, but not so well for our taxa. Assuming there is no bias, what are the forces that drive the GC-content variation within and between our genomes? And how is it that recombinogenic parts of the genome in three of our four species are GC rich? We are looking forward to see how further studies answer these questions.
Haoxuan Liu, Sihai Yang, and Laurence D. Hurst
Liu H, Huang J, Sun X, Li J, Hu Y, Yu L, Liti G, Tian D, Hurst LD, Yang S (2017) Tetrad analysis in plants and fungi finds large differences in gene conversion rates but no GC bias. Nature Ecology & Evolution 10.1038/s41559-017-0372-7




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in