Dose matters: Understanding adaptation of Hepatitis C virus to cell culture
Published in Microbiology
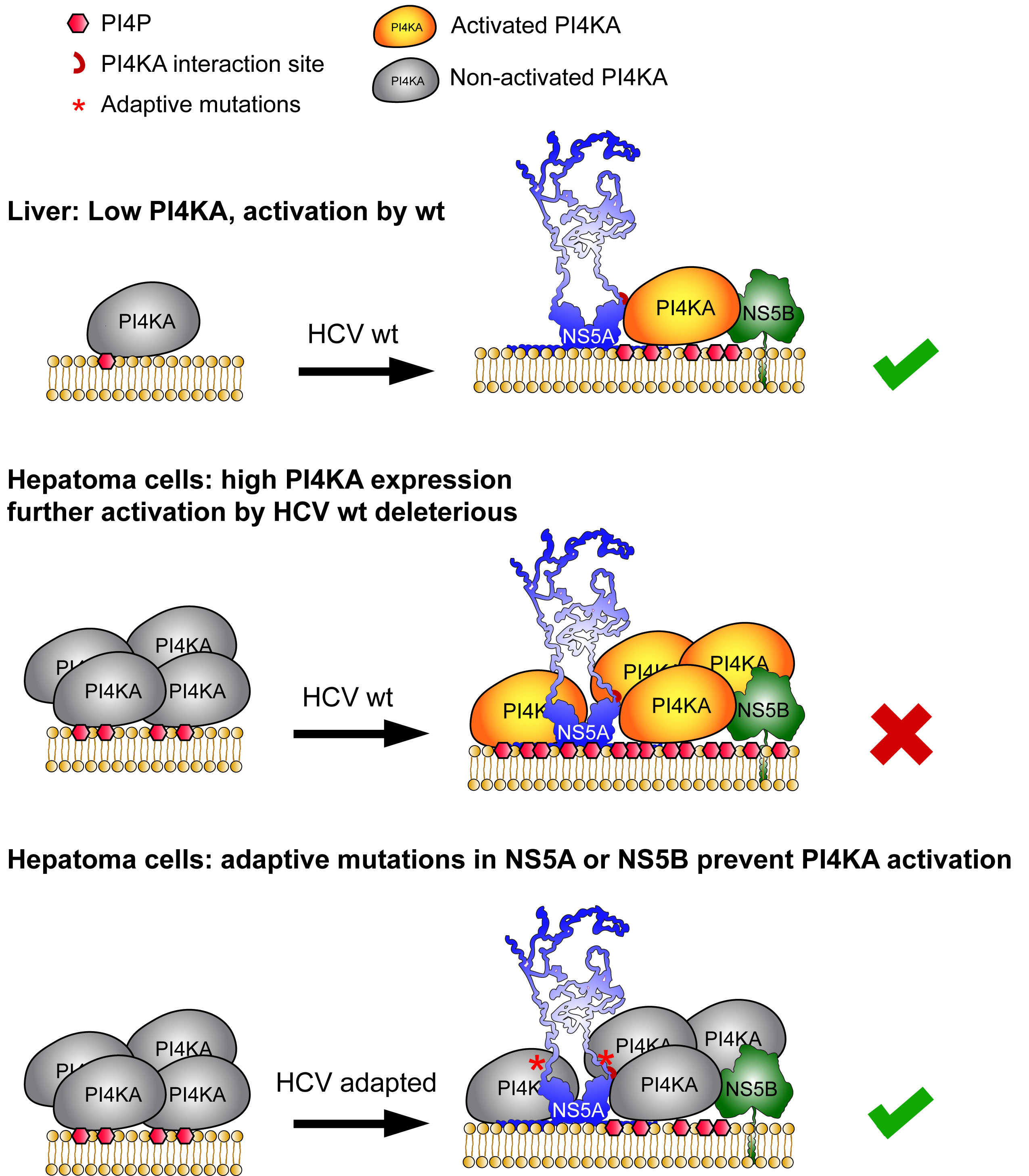
This paper bridges almost my entire scientific life. I started my PhD in 1993 and spend several frustrating years on the development of a cell culture model for Hepatitis C virus (HCV). In 1999, during my first year as a postdoc we finally succeeded by creating a so called subgenomic replicon, encoding only the proteins required for viral genome replication and a selectable marker gene. With this strategy we selected a few resistant cell clones with at that time unbelievably high levels of HCV replication. It took us again years to understand the reasons for our success. First, we were extremely lucky in picking the right viral genome, a genotype 1b isolate termed Con1. Second, we were lucky enough to include the right cell line, Huh-7 hepatoma cells, which is still the cell type with highest permissiveness for HCV replication. Third the viral genome had acquired adaptive, replication enhancing mutations, mainly located in the viral nonstructural proteins NS5A and NS5B, dramatically increasing the efficiency of the system. At that time we had no clue on their mode of action.
Years later, an siRNA screen brought our attention to the lipid kinase Phosphatidylinositol 4-kinase IIIα (PI4KA), converting phosphatidylinositol (PI) to PI 4-phosphate (PI4P), which was identified by us and others as an essential host factor of HCV replication. We found that HCV had established a way to stimulate PI4KA activity via the viral nonstructural proteins NS5A and NS5B, resulting in dramatically increased PI4P levels in vitro and in vivo. The PI4KA interaction site in NS5A was very close to a hotspot of adaptive mutations; therefore, we thought about a functional link between PI4KA and cell culture adaptation. Indeed, we found to our own surprise that all adaptive mutations in NS5A and NS5B abrogated PI4KA activation, thereby representing loss-of-function mutations! The explanation was in the end rather simple: While in the liver, the natural environment of HCV, PI4KA expression levels are rather low, the virus has evolved a mechanism to activate this lipid kinase, to reach PI4P levels sufficient for efficient replication. However, PI4KA expression levels are increased in liver tumors and consequently are elevated in hepatoma cells like Huh-7. Under conditions of high PI4KA expression, further activation of PI4KA results in an overproduction of PI4P, which is deleterious for replication. Therefore, the virus needs to adapt by tuning done activation of PI4KA. Using a special PI4KA-inhibition regimen we are now able to replicate wildtype-virus from patients in cell culture, which has been a dream since I started working on HCV. Apart from this, our paper has important take-home-messages beyond HCV: 1. Most viral cell culture models are based on cell lines established from tumors and we tend to underestimate the global changes in gene expression associated with tumorigenesis. 2. As a consequence, many viruses have to adapt to this in vitro environment, but we typically do not care about the underlying mechanisms. 3. If a couple of diverse mutations cause the same complex phenotype (e.g. adaptation) it is far more likely that this is mediated by a loss-of-function that by a gain-of-function. 4. We often think of host factors in black or white. However, as we show here, it can be a matter of dose turning an essential host factor into too much of a good thing, restricting viral replication. Think of that next time, when your host factors switch between pro- and anti-viral in different experimental settings.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in