Efficient Machine Learning Protocol For Photodynamics Simulations
Published in Chemistry and Computational Sciences
Quantum chemical simulations of molecular excited states are important for understanding the photochemical and photophysical processes, but they are very computationally intensive, especially when used for nonadiabatic dynamics. Machine learning is an attractive tool to tackle this problem by replacing expensive quantum chemical calculations, however, the progress in ML for excited states is much slower than for ground states due to the complexity of the former (see, e.g., our review in Nat. Rev. Chem. 2021, 5, 388–405).

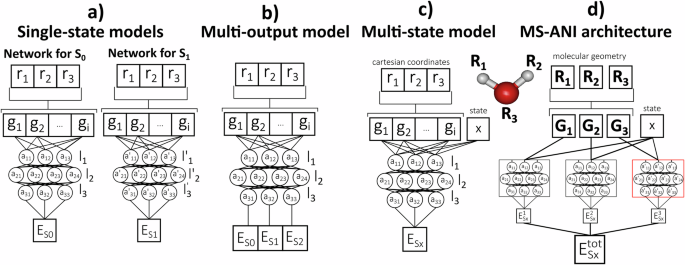
To address the key challenges in machine learning of excited states, this work has reported a comprehensive and robust protocol for ML based on an eight-year-long (!) in-depth research. This protocol is based on a multi-state learning model, MS-ANI, capable of learning excited states of various molecules with unprecedented accuracy. The core idea is to take the state number as an input feature, concatenated with the ANI atomic environment vectors, and passed to the neural network for the corresponding element type. Impressively, the accuracy of learning excited states is better or similar to the accuracy of learning ground-state energies, where information on excited-state energies improves the quality of ground-state predictions.

One of the key ingredients to train a robust potential for nonadiabatic dynamics is accelerated sampling of the regions near conical intersections. We have achieved this in this work by introducing gapMD, which ensures enhanced sampling of the low-gap regions by propagating trajectories with the gradient energy difference between adjacent surfaces when needed.

Together, these advances enabled end-to-end active learning for generating machine learning models to perform nonadiabatic simulations.

For simple cases such as fulvene, this active learning is very fast and converges just after three days on a single GPU card.

Active learning also works for more complex cases and, e.g., can be used to calculate photoreaction quantum yields. We have also discovered long-time-scale oscillations in cis-azobenzene photoisomerization.

In this work, we have also shown for the first time that a single model can be used to perform the nonadiabatic dynamics of different molecules. This has laid the foundation for our follow-up study on the first-ever universal excited-state potential OMNI-P2x.
All the methods, together with the end-to-end active learning, are available in MLatom. The open-source code and tutorials: https://github.com/dralgroup/al-namd.
Video lecture about the theory and a life-recorded tutorial: https://www.youtube.com/watch?v=_9-v-VZaoo0.
Professor in Xiamen University and Nicolaus Copernicus University in Toruń; Aitomistic, Shenzhen. Artificial intelligence for atomistic simulations.
Follow the Topic
-
npj Computational Materials
This journal publishes high-quality research papers that apply computational approaches for the design of new materials, and for enhancing our understanding of existing ones.

Related Collections
With Collections, you can get published faster and increase your visibility.
Altermagnetic Materials: Theory, Simulation, and Renewed Perspectives
Publishing Model: Open Access
Deadline: Feb 28, 2027
Recent Advances in Active Matter
Publishing Model: Open Access
Deadline: Sep 01, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in