Epigenetic engineering for oncotherapy
Published in Bioengineering & Biotechnology

After taking a closer look at epimutations underlying the origin and progression of cancer on a very recent post here, followed by a brief experimental strategy to examine oncogenesis with cancer stem cells. I continue to explore my cross-disciplinary fascination on a similar theme – by focusing on epigenetic therapy. In previous works, biologists had likened epigenetic changes that occur during oncogenesis to biological switches that can induce reversible DNA modifications, which can be turned on or off – without changing the sequence of the DNA building blocks [Dawson 2017, Waddington 1942].
Beyond the 'mainstream' search for proximate causes that underly cancer, there is an aim to explore the disease as a biological phenomenon that results from the progressive erosion of the epigenetic landscape (Figure 1). This has resulted in attempts to target the products or effects of driver mutations as a leading strategy to achieve therapeutic control of cancer [Aranda-Anzaldo 2018]. In this post, I briefly examine the scope of epigenetic therapy in the field of immune oncology to reverse epigenetic modifications for cancer treatment and management. Broadly speaking, epigenetic engineering aims to reshape the tumor microenvironment and reprogram immune cells. Therapeutic interventions can be juxtaposed with varied epigenetic mutations, to gain further insights to the evolution of several disease signaling pathways.
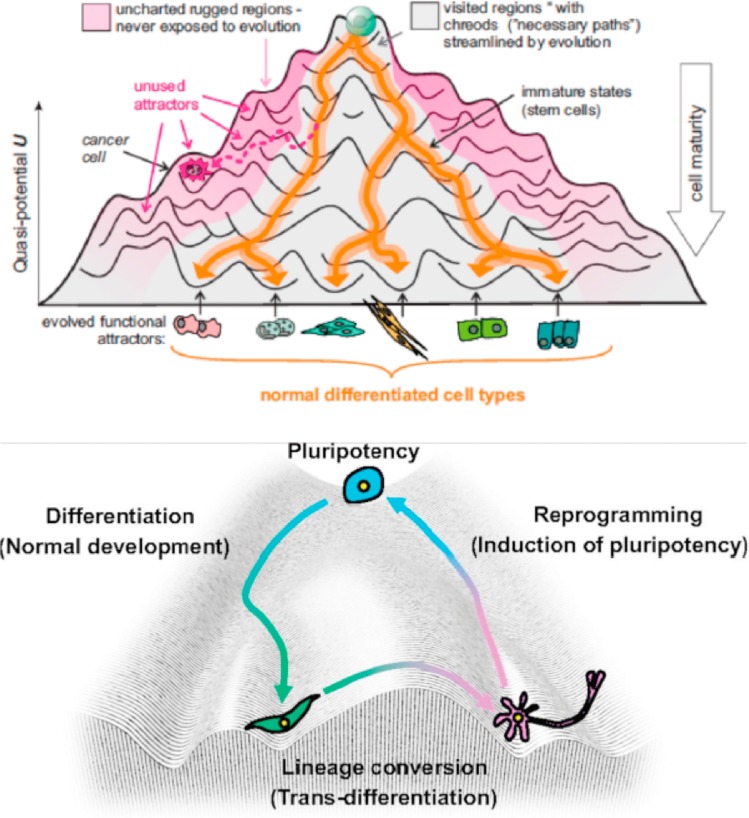
Figure 1: The epigenetic landscape. Waddington’s epigenetic landscape simplified the complexity of Metazoan development and differentiation. An alternative emerging perspective that understands cancer as a tissue/organ morphology and structural organization-related concept is receiving greater attention [Aranda-Anzaldo 2018].
Writers, readers, and editors of the epigenome
Drugs engineered for epigenetic regulation of solid tumors depend on targeting four factors: writer, eraser, reader, and remodeler-associated enzymes of the tumor microenvironment [Jin 2021]. Enzymes that add acetyl or methyl groups to the histone or DNA are ‘writers,’ while enzymes associated with the removal of histone marks are ‘erasers,’ and those that recognize histone and DNA modifications are chromatin ‘readers’ [Arrowsmith 2012] (Figure 2). In cancer research, scientists tread a fine balance to delineate normal and abnormal epigenetic features to effectively develop therapies.
Clinical trials classify epigenetic drugs into two major classes; reprogrammers (broad spectrum) and target therapies (narrow spectrum) specific to treat patient subsets [Jones 2016]. During cancer progression, epigenetic regulations can arise via DNA methylation (writers), to silence tumor suppressor genes. It is therefore a rationale treatment strategy to develop DNA methylation inhibitors (DNMTi) that target specific types of hematological malignancies [Jones 2019]. In its mechanism of action in general, during DNA methylation, cytosine is methylated and converted to 5-methylcytosine. During hypermethylation-induced transitional mutations, methylation inhibitors act as cytidine analogs to induce loss of DNA methylation [Issa 2009].
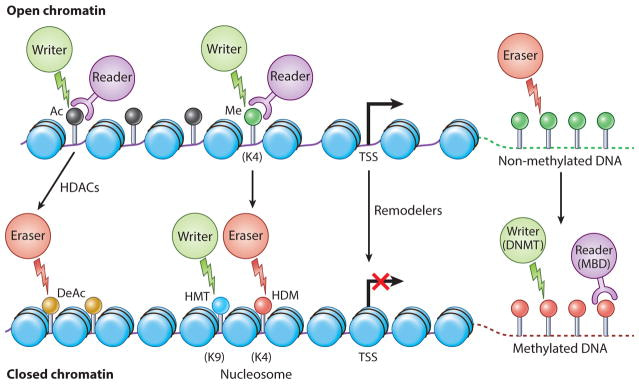
Figure 2: The four Rs of epigenetic protein regulation. Epigenetic signal writers (green circles), readers (purple circles) and erasers (red circles). Generally, no DNA methylation is associated with CPG islands (green lollipops) [Ahuja 2016].
The DNMTi agents can also address the problem of cytotoxic T lymphocyte exhaustion in the tumor microenvironment to return exhausted immune cell types back to their original phenotypes, for an enhanced anti-tumor immune response. To emphasize the significance of this mechanism-of-action of DNMTi agents, existing modes of immune checkpoint blockade therapies including anti-programmed cell death ligands cannot fully reprogram such exhausted immune cells back to effector phenotypes [Jones 2019][Ghoneim 2017].
The evolution of therapeutic strategies
The treatment options of epigenetic engineering can be briefly classified to four main categories by juxtaposing mutational events and epigenetic alterations (figure 3). The regimen of drugs is evolving with rapidly evolving mutations. While therapeutic intervention can be implemented primarily with DNA methylation inhibitors alone, where significant alterations underlying methylomes in human cancers have made DNMTis suited for myelodysplastic syndromes and acute myeloid leukemia in the form of monotherapies – not all patients benefit from monotherapy [Issa 2009].
DNMTi agents and histone deacetylase ‘eraser’ inhibitors (HDACi) together can cause changes in the epigenome in general [Ahuja 2016] as combined therapy. The next phases of treatment strategies include DNMTis combined with immunotherapy, chemo, and targeted therapy. The fourth phase integrates readers, writers, and small molecules, of which small molecules are now being introduced in phase I clinical trials [Tanaka 2015].
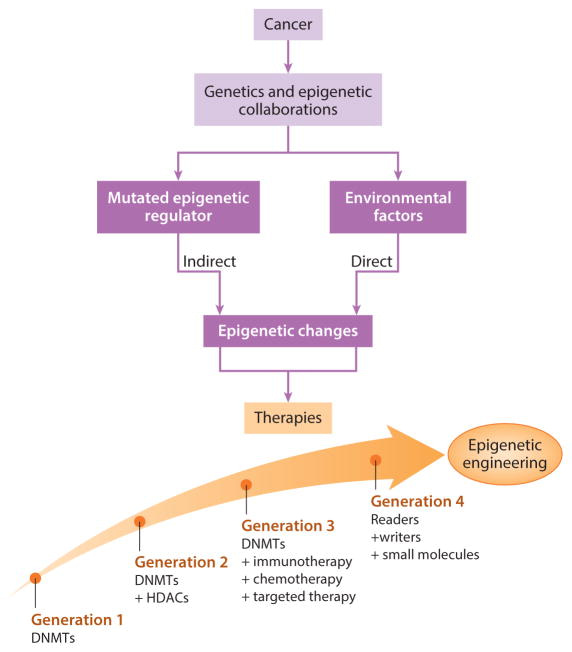
Figure 3: The evolution of epigenetic engineering. Treatment options can juxtapose mutational events and epigenetic alterations. Cancers effect the epigenome directly to result in chronic inflammation, viral infections, or microbiome changes as a result of mutations in epigenetic driver genes. Based on the outcomes of preliminary phase I clinical trials, the regimens of epigenetic therapy can be optimized to facilitate the next-generation of evolving therapies [Ahuja 2016].
A few broad spectrum reprogrammers.
Histone deacetylases that remove acetyl groups on histones are erasers, they can make the chromatin condensed and transcriptionally silent, and are key regulators of gene expression, differentiation, and development for cellular homeostasis maintenance. During tumorigenesis, the HDAC inhibitors (HDACi) can upregulate global genes such as onco-repressors and cause tumor growth arrest by inhibiting oncogenes [Greer 2015]. A variety of these drugs, including DNMTi agents, were FDA approved in clinical trials [Jones 2016].
Cancer biologists are also excited about chromatin-associated proteins such as bromodomain and extraterminal motif proteins (reader) and their associated inhibitors (BET inhibitors) that can recognize and regulate hyperacetylated nucleosomes, to downregulate specific genes and induce anti-leukemic effects associated with acute myeloid leukemia [Jin 2021]. These inhibitors can also selectively repress transcriptional networks driven by the c-Myc gene; a master regulator activated by several oncogenic pathways [Delmore 2011, Miller 2015]. Additionally, CDK9 inhibitors are associated with gene expression alterations in acute myeloid leukemia too [Boffo 2018], they target the cyclin-dependent kinase 9 pathway that is dysregulated during oncogenesis.
RNA epigenetic markers
Aside from DNA or histone modifications, eukaryotic RNAs can also undergo methylation such as N6-methyladenosine (m6A) modification of messenger RNA (mRNA). This modification is installed by a multiprotein writer complex containing methyltransferase-like protein 3 (METTL3) and other accessory subunits. These epigenetic modifications are upregulated in diverse cancer types, including acute myeloid leukemia, renal cell carcinoma, non-small cell lung cancer and gastric cancer [Vu 2017]. Several inhibitors of METTL3 are being studied in leukemia and solid tumors, with pending clinical trials [Cully 2019].
Narrowed spectrum reprogrammers - Targeted therapies to treat specific genetic alterations.
Of the target therapies, narrowed spectrum reprogrammers can address isocitrate dehydrogenase (IDH) mutations that occur during acute myeloid leukemia, and glioblastoma. Mutations of IDH usually block the production of alpha-ketoglutarate; a core factor underlying multiple proteins that regulate the epigenome. Small molecules targeting IDH mutations are in clinical trials with early promise to treat acute myeloid leukemias [Ahuja 2016]. Similarly, the enhancer of zeste homologue 2 (EZH2) gain-of-function mutations in lymphomas can be regulated by EZH2 inhibitors.
Additional reprogrammers include lysine-specific histone demethylase 1 ‘erasers’ that can erase monomethyl and dimethyl chromatin marks on histone and act as transcriptional repressors or activators, overexpressed in a variety of solid tumors, including colorectal cancers and neuroblastoma. Their pharmacological inhibition offers promising potential therapeutic options in a variety of cancer types. Similarly, the disruptor of telomeric silencing 1-like gene inhibitors, are a new therapeutic target specific to epithelial-to-mesenchymal transitions to treat aggressive breast cancer [Lee 2015]. The current FDA-approved epigenetic therapies to treat malignancies are presented on table 1.
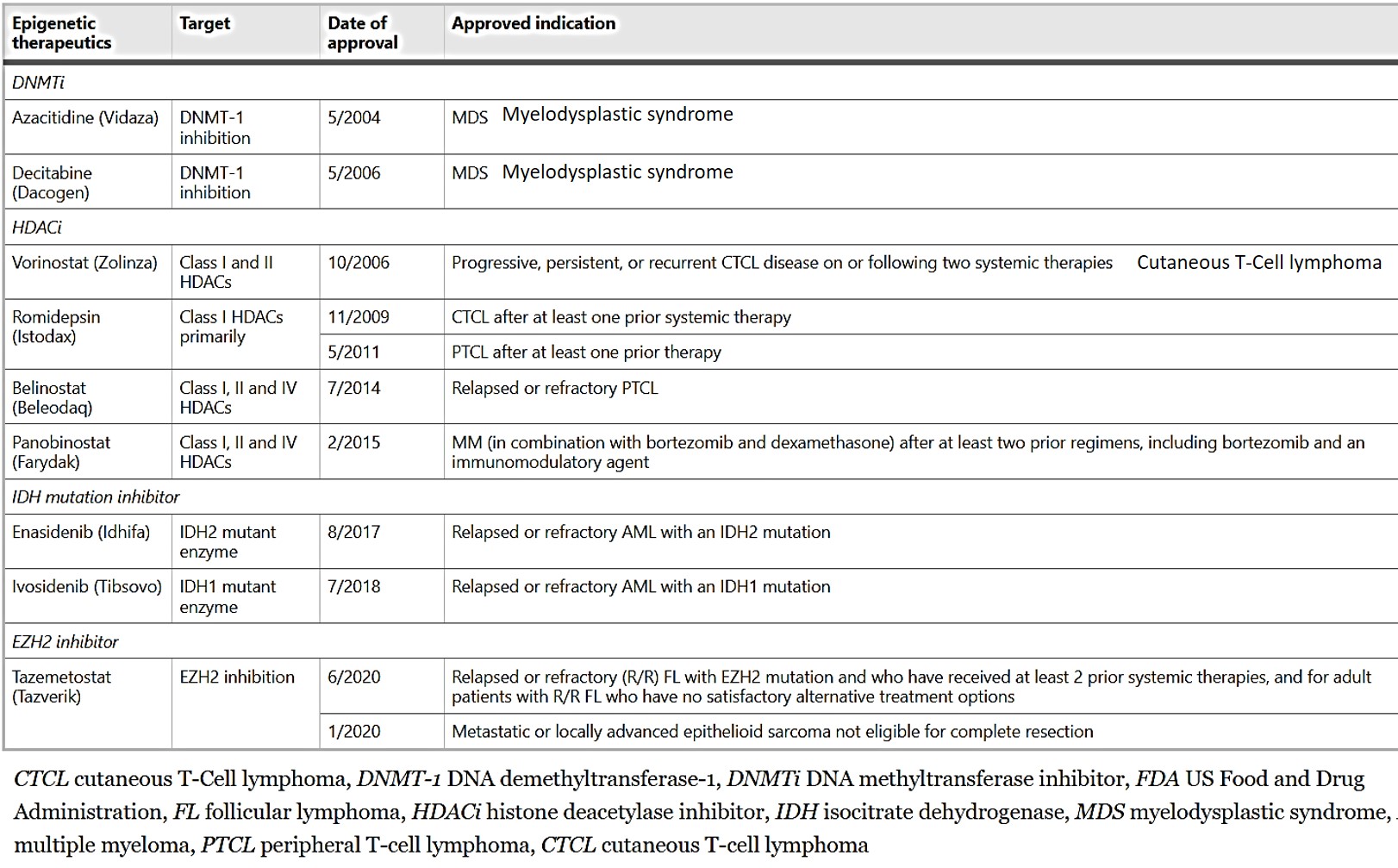
Table 1: FDA-approved epigenetic therapeutics in malignancies [Jin 2021].
Principles of immune checkpoint inhibition and a viral theory
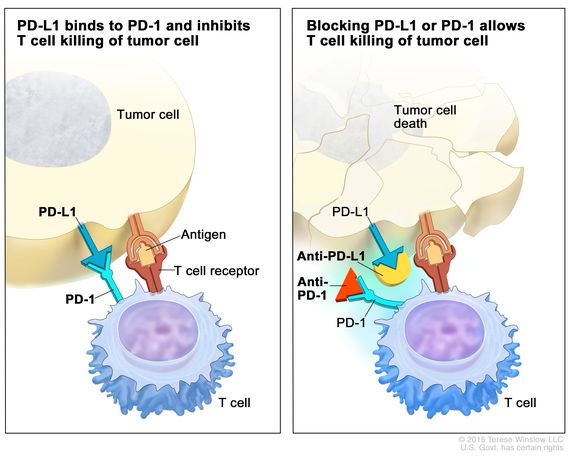
In a nutshell, immune checkpoints arise during cancer progression to engage on T-cells by using ligands expressed by tumors, or by immunosuppressive cellular populations in the tumor microenvironment. This process weakens the host’s immune response and attenuates T-cells from killing cancer cells. Immune checkpoint inhibitors are a drug that blocks such checkpoint inhibitors, to increase the capacity of T cells to kill the cancer cells (Figure 4) [Jones 2019].
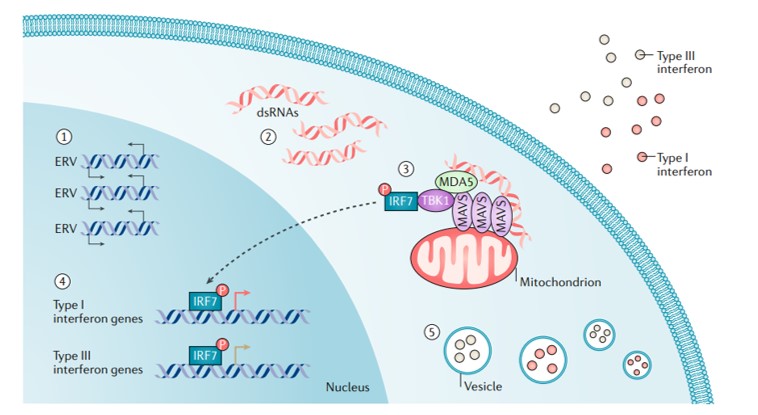
It is also known that several endogenous retroviruses, transposable elements, and long-interspersed elements can be activated during epigenetic treatment strategies to induce a state of viral mimicry. In this scenario, treated cancer cells interpret the treatment as an infection caused by an exogenous virus to mount an immune response that leads to the production of type I, III, interferons to elicit an anti-inflammatory response [Chiappinelli 2015] (Figure 5).
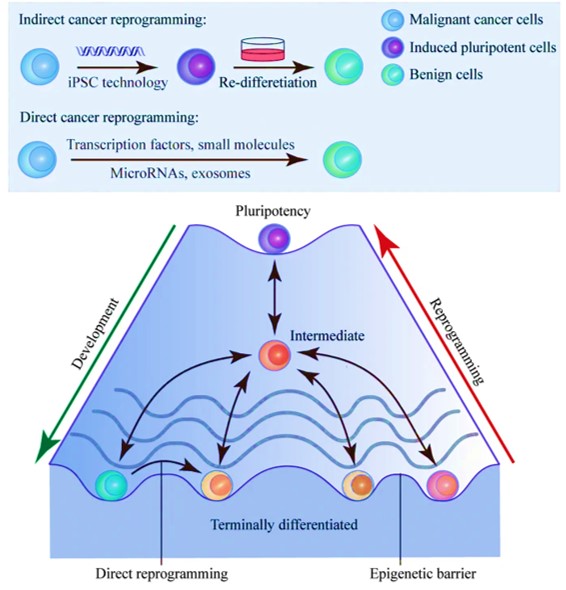
Another promising cancer therapy strategy that is still in development can reprogram malignant cells to benignity [Gong 2019]. The concept relies on the inherent cellular plasticity that can reprogram terminally differentiated somatic cells to other lineages. Based on the same principles, the advent of transcription-factor mediated cancer cell reprogramming is a groundbreaking game-changer that yet remains to enter clinical trials (Figure 6).
Working together – combined therapies reverse acquired chemoresistance.
To wrap things up, epigenetic monotherapy is a complex process that sometimes necessitates combinatorial therapy in solid tumors. These include the combination of epigenetic modifiers with agents for cytotoxic chemotherapy, hormonal therapies, and as immune checkpoint inhibitors. For example, preclinical studies have shown the significance of combining DNA methyl transferase inhibitors (DNMTis) and histone deacetylase inhibitors (HDACis) to increase apoptosis in cancer cells [Zhu 2001].
These combinative epigenetic therapies can be merged with cytotoxic chemotherapy with great efficiency to re-sensitize cancers to standard cytotoxic agents [Benson 2015]. For example, clinical studies have shown that the acquired resistance of chemotherapy can be reversed when combined with DNMTis, and/or HDAC inhibitors during ovarian cancer [Sharma 2010]. Several other epigenetic therapies can be administered with anticancer therapies, to overcome treatment resistance. Ongoing clinical trials of combined anticancer therapies and their strategies of targeting cancer are listed herein [Jin 2021].
Outlook – seeing the bigger picture at the molecular level.
This post only highlights a brief snippet of epigenetic engineering strategies that attenuate tumorigenesis. Cancer researchers strike a fine balance to delineate between normal vs. abnormal epigenetic features to develop personalized strategies for medicine and diagnostics that reshape the cancer microenvironment and reprogram immune/cancer cells. This article brings together a series of epigenetic engineering strategies to address epigenetic mutations underlying cancer progression by developing precision and personalized diagnostics/therapies, some of which are FDA approved while others are still being explored in clinical trials.
Based on the outcomes of preliminary phase I clinical trials, the regimens of epigenetic therapy can be optimized to facilitate the next generation of evolving treatment options, of which some can induce viral mimicry in the tumor microenvironment, to kickstart an antiviral response that targets cancer cells. While others can reprogram malignant cells to benignity [Gong 2019].
From an investigational viewpoint, studying the evolving mechanism-of-action behind drug-immune cell interactions can provide fascinating insights to further understand the evolution of pathophysiological pathways, and explore multiple molecular mechanisms behind the origin and progression of different cancer types. This is further emphasized by classifying treatment options after juxtaposing mutational events with epigenetic alterations within the ever-changing epigenetic landscape during cancer progression, to examine and enable continued investigations on drug discovery and basic science discovery in cancer medicine.
Header Image: Fundamental mechanisms of epigenetic gene regulation, Credit: PubMed, doi: 10.1152/japplphysiol.00131.2010
References
- Dawson M., The cancer epigenome: Concepts, challenges, and therapeutic opportunities, Science, 2017, doi: 10.1126/science.aam7304
- Waddington C., Canalization of development and the inheritance of acquired characteristics. Nature, 1942, doi: https://doi.org/10.1038/150563a0
- Aranda-Anzaldo, A. et al., Landscaping the epigenetic landscape of cancer, Progress in Biophysics and Molecular Biology, 2018, doi: https://doi.org/10.1016/j.pbiomolbio.2018.06.005
- Jin N. et al. Advances in epigenetic therapeutics with focus on solid tumors. Clinical Epigenetics, 2021, doi: https://doi.org/10.1186/s13148-021-01069-7
- Arrowsmith C.H. et al. Epigenetic protein families: a new frontier for drug discovery, Nature Reviews Drug Discovery, 2012, doi:10.1038/nrd3674
- Jones P. et al. Targeting the cancer epigenome for therapy, Nature Reviews Genetics, 2016, doi: https://doi.org/10.1038/nrg.2016.93
- Jones P. et al. Epigenetic therapy in immune-oncology, Nature Reviews Cancer, 2019, doi: https://doi.org/10.1038/s41568-019-0109-9
- Issa J. et al. Targeting DNA methylation, Clinical Cancer Research, 2009, doi: 10.1158/1078-0432.CCR-08-2783
- Ghonheim H. et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation, Cell, 2017, doi: 1016/j.cell.2017.06.007
- Ahuja N. et al. Epigenetic Therapeutics: A New Weapon in the War Against Cancer, Annual Review of Medicine, 2016, doi:10.1146/annurev-med-111314-035900
- Tanaka M. et al. Inhibitors of emerging epigenetic targets for cancer therapy: a patent review (2010-2014), Pharmaceutical Patent Analyst, 2015, doi: 10.4155/ppa.15.16
- Greer C. et al. Histone Deacetylases Positively Regulate Transcription through the Elongation Machinery, Cell Reports, 2015, doi: 10.1016/j.celrep.2015.10.013
- Miller D. et al. c-Myc and Cancer Metabolism, Clinical Cancer Research, 2015, doi: 1158/1078-0432.CCR-12-0977
- Delmore J. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc, Cell, 2011, doi: 10.1016/j.cell.2011.08.017
- Boffo S. et al. CDK9 inhibitors in acute myeloid leukemia, Journal of Experimental and Clinical Cancer Research, 2018, doi: 10.1186/s13046-018-0704-8
- Vu L. et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells, Nature Medicine, 2017, doi: 10.1038/nm.4416
- Cully M. Chemical inhibitors make their RNA epigenetic mark, Nature Reviews Drug Discovery, 2019, doi: 10.1038/d41573-019-00179-5
- Lee J. et al. DOT1L: a new therapeutic target for aggressive breast cancer, Oncotarget, 2015, doi: 10.18632/oncotarget.5860
- Zhu W. et al. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors, Cancer Research, 2001 doi: https://pubmed.ncbi.nlm.nih.gov/11245429/
- Benson E. et al. Carboplatin with Decitabine Therapy, in Recurrent Platinum Resistant Ovarian Cancer, Alters Circulating miRNAs Concentrations: A Pilot Study, PLoS One, 2015, doi: 10.1371/journal.pone.0141279.
- Sharma S. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations, Cell, 2010, doi: 10.1016/j.cell.2010.02.027.
- Chiappinelli K. et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses, Cell 2015, doi: 10.1016/j.cell.2015.07.011.
- Gong L. et al. Cancer cell reprogramming: a promising therapy converting malignancy to benignity, Cancer Communications, 2019, doi: https://doi.org/10.1186/s40880-019-0393-5
I am an interdisciplinary researcher with a strong commitment to bioengineering, biochemistry, organ-chips and molecular biology. As well as biomechanics and biomineralization in the broader context of medicine. I completed my PhD at the University of Sydney Australia in December 2016, and travel often, find me on Twitter and irl.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in