Experimental biology – Bioinspired engineering on a kidney chip to investigate renal calcification
Published in Bioengineering & Biotechnology, Biomedical Research, and General & Internal Medicine
In this post I revisit a recurrent (and therefore a favorite) plot of bottom-up bioengineering a pathological pathway of renal calcification. I previously focused on this topic, briefly, to identify an oxidative-stress related biological switch at the renal tip and then defined the influence of machine learning and systems biology within the niche, followed by a more recent post on structural biology or basic science, to recreate bioinspired mechanobiology-on-a-chip or on a microfluidic platform. Aspects of the following work were accepted as an abstract for publication at the World Congress of Nephrology in January 2023.
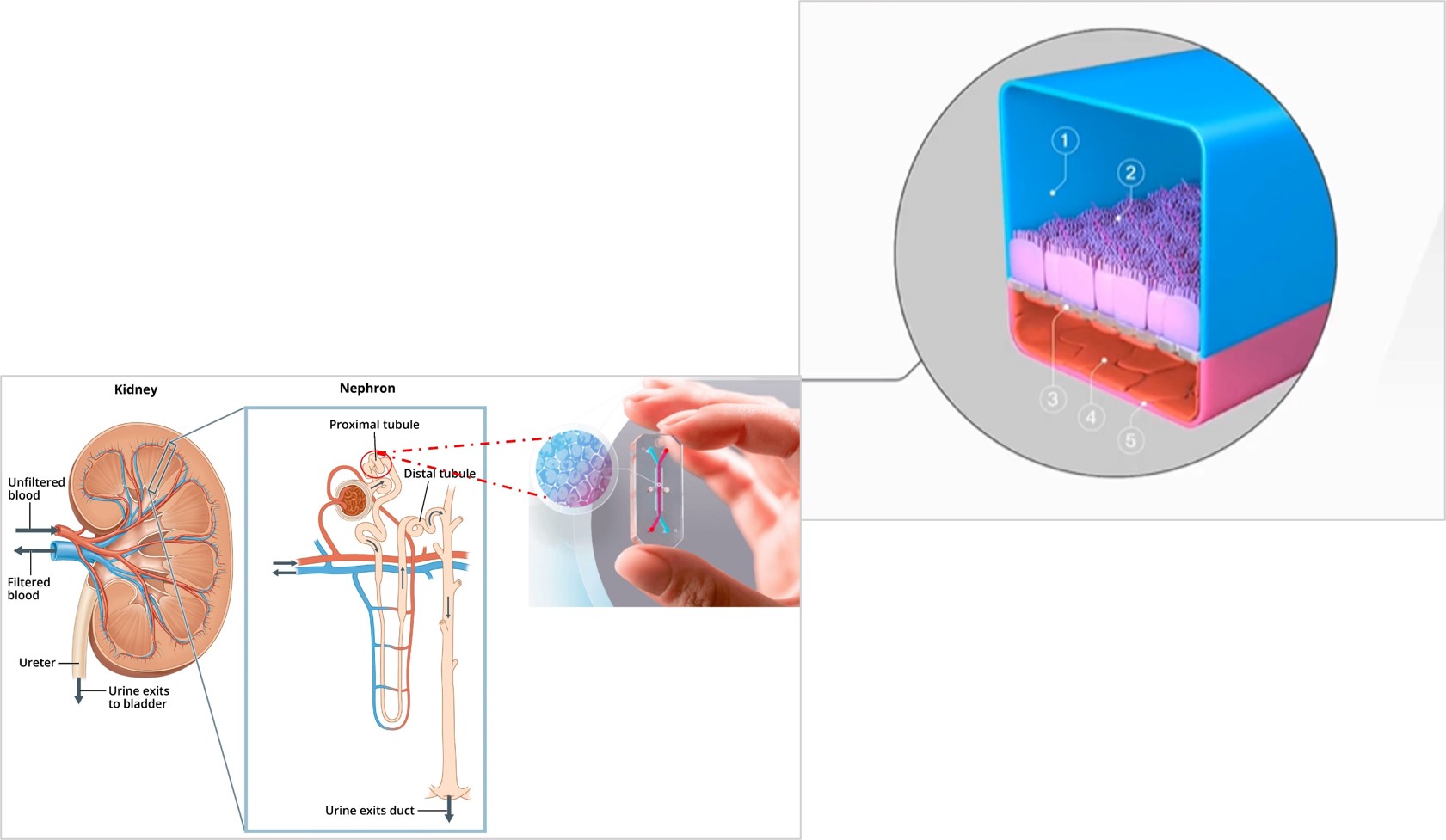
Our primary aim is to understand the influence of mechanosensitive ion channels and their downstream triggers during renal calculi formation (Figure 1). The secondary aim is to isolate a regulatory mechanism of an oxidative stress pathway. The kidney-on-a-chip platform offers a microphysiological environment in vitro to distinctly recreate representative regions of the medullary pyramid of the kidneys, consisting of the nephrons, proximal tubules, glomerular epithelium, and ureteric epithelium.
This post answers a few questions that I posed earlier, with supportive first-in-study outcomes in tow:
- Is there a biological switch at the renal tip that triggers the onset of calcification?
- Does the mechanotransduction ion channel Piezo1 activate the upregulation of renal fibrosis during calcification and contribute to kidney stone formation?
- Can identifying the culprit proteins and regulatory biological mechanisms, offer a potential target of pharmacotherapy to attenuate kidney disease?
In response, I present a series of foundational basic science experiments as a basis to investigate pathological cascades via bioinspired engineering. And then I briefly bring together first-in-study outcomes completed in-lab as a research lead at the University of California San Francisco, to bottom-up engineer a pathological pathway of renal calcification - as discussed in detail within the context.
A prelude to endothelial mechanotransduction and renal injury
Despite a variety of flow-sensitive mechanisms that affect endothelial cells, the process of endothelial mechanotransduction during urolithiasis or nephrolithiasis (renal stone formation) remains to be known in depth. Endothelial dysfunction is an identified trigger of multiple co-morbidities such as high blood pressure, diabetes, and vascular disorders, with a specific role during lithogenesis or calculi (stone) formation [Saenz-Medina 2022]. Alongside flow-mediated mechanotransduction, the process of endothelial dysfunction in urolithiasis and nephrolithiasis appears to harness reactive oxygen species as an underlying biomechanism [Aydin 2010, Saenz-Medina 2021].
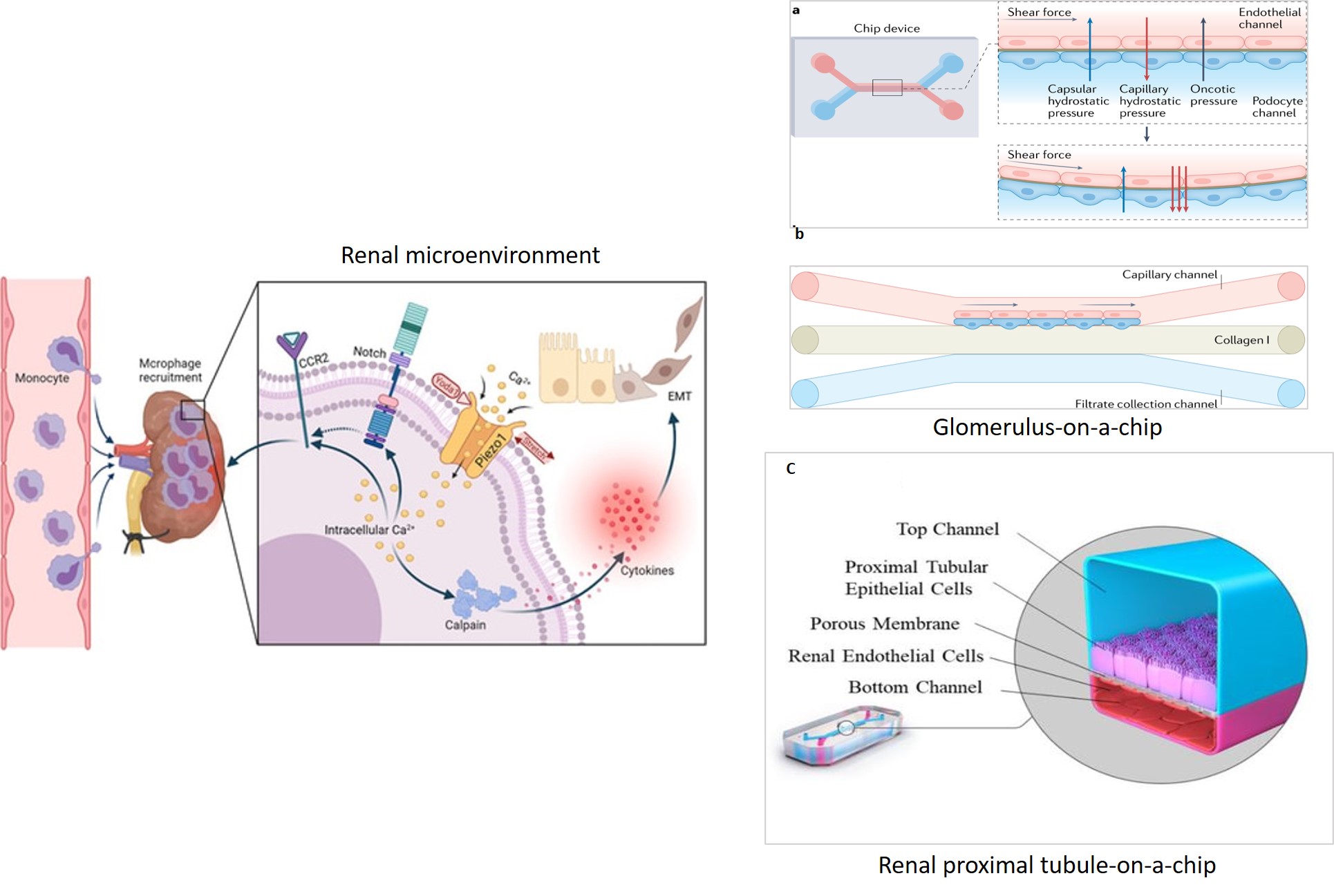
Here, I look at the experimental basis of the combined effects of reactive oxygen species, Piezo1 and the transient receptor potential vanilloid subfamily 4 (TRPV4) mechanosensory ion channels involved in downstream calcium signaling, to assess endothelial dysfunction and fibrosis. Tissue fibrosis is a precursor of disease and an intermediate clinical feature seen at the onset of calculi formation and urolithiasis. In its mechanism-of-action, shear-stress related increase in mechanotransduction converts physical forces into biochemical signals, to trigger tissue injury that eventually leads to progressive renal calcification (Figure 2) [He 2022]. Kidney-on-a-chip models can be created without using artificial materials to simulate the filtration membrane, where the extracellular matrix is developed using collagen and Matrigel protein to facilitate cell attachment and proliferation in renal microphysiological environments recreated on an organ-chip instrument, to represent pathology (Figure 2).
A snapshot of the bigger picture – flow mediated endothelial mechanotransduction in renal fibrosis
The present focus on Piezo1 and TRPV4 mechano-active ion channels bring us back to the tour-de-force discoveries of Patapoutian et al., [Coste 2010] whose emphasis on unveiling the combined mechanisms of TRPV1 and Piezo mechanosensory ion channels led to the 2021 Nobel prize in physiology and medicine. Such groundbreaking studies in structural biology and basic science have shed light on the channel gating mechanisms in plasma membranes of neuronal cells at the atomic level, to reveal receptors for ‘temperature and touch,’ for the first time [Coste 2010, Ranade 2015, Cheng 2022].
Much like Piezo1, endothelial cell-expressed calcium transient receptor potential vanilloid subfamily 4 (hitherto referred to as TRPV4) channel proteins are expressed in a variety of pressure-sensitive cells and tissues and are composed as tetramers of identical subunits (Figure 3) [Deng 2018]. Biomechanically, Piezo1 acts upstream of TRPV4 to induce pathological changes primarily in epithelial, endothelial, and smooth muscle cells in response to shear stress [Swain 2021]. The strength and duration of fluid shear can impact the upregulation of Piezo1 subunits – to increase TRPV4 channel activation and elevate intracellular calcium levels for both physiologic and pathologic outcomes [Dalal 2020].
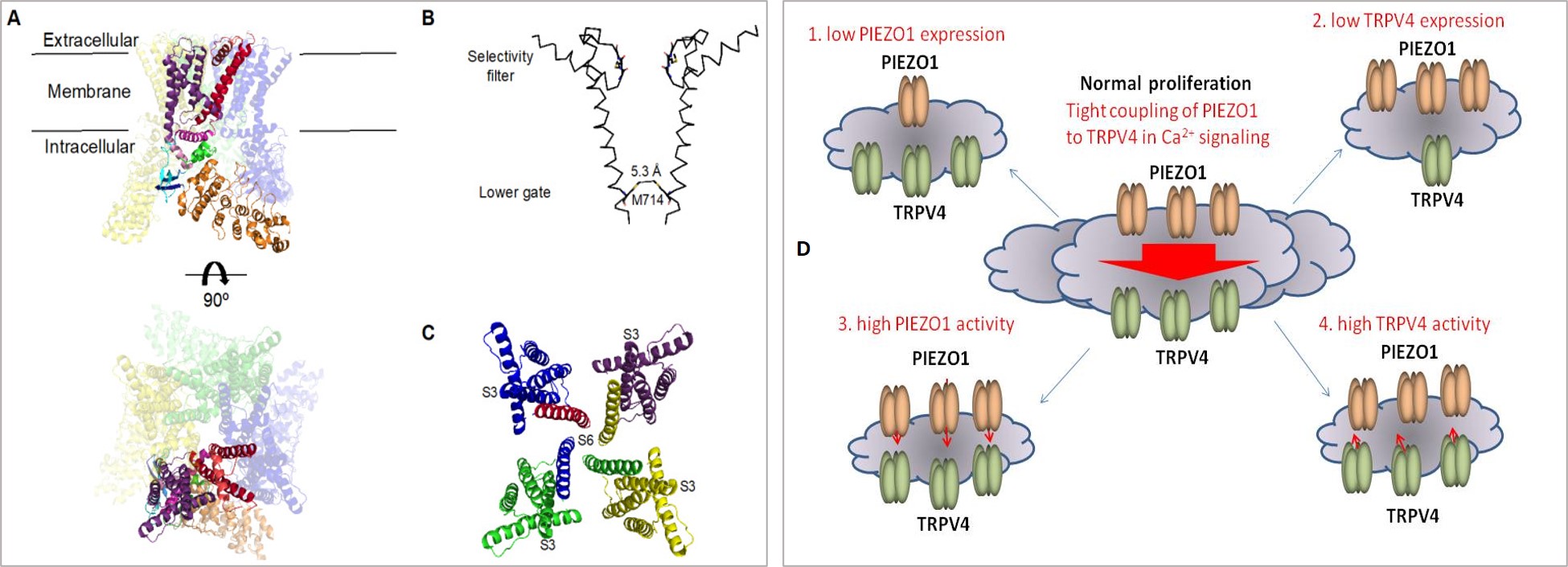
Figure 3: Cryo-EM structure of TRPV4. A) Cryo-EM reconstruction of the tetrameric channel, b) orthogonal views of the overall structure, c) structure of a single subunit, d) schematic representation of the Piezo1 and TRPV4 interaction dynamics during mechanosensory ion channel transduction involved at downstream calcium signaling. Credit: [Yoneda 2019, Deng 2018].
Although TRPV4 is functionally similar to Piezo1, TRPV4 proteins do not have true mechanoreceptor properties, and therefore their capacity to sense physical force in endothelial cells is not fully understood [Mochizuki 2009, Strotmann 2000]. It is assumed that Piezo1 can stimulate the TRPV4 channel protein opening by activating phospholipase A2. TRPV4 is linked to several physiological functions including epithelial ciliary activity and blood flow regulation, to then play a pathological role in actin disruption in response to shear stress – as seen during kidney fibrosis [Mendoza 2010]. Pharmaceutically, it is possible to circumvent the associated loss of endothelial cell integrity by blocking TRPV4 via its antagonist drug HC067 [Li 2021, Everaerts 2010].
The running postulation is therefore that pressure-induced mechanotransduction driven by Piezo1 signaling coupled to TRPV4 activation can account for the impact of shear stress on epithelial and endothelial cells, leading to a sustained elevation of calcium ions and a loss of endothelial contacts, to cause actin disruption and endothelial dysfunction during vascular disease and nephrolithiasis [Swain 2021].
Basic science – a few contenders for renal calculi generation: ROS, Piezo1, TRPV4
Stone formation is hypothetically yet another case of pathological biomineralization and there is much to learn from osteogenesis and atherosclerosis [Gambaro 2004, Khan 2021]. Nephrolithiasis is a global health issue that can proceed to chronic kidney disease in most severe cases, although no single theory has clearly established kidney stone formation on account of the variety and complexity of its occurrence [Duffield 2014, Coe 2010]. Epithelial injury and endothelial dysfunction are clinical intermediates of urolithiasis and nephrolithiasis, potentially driven by pathological intracellular mechanotransduction [Saenz-Medina 2022].
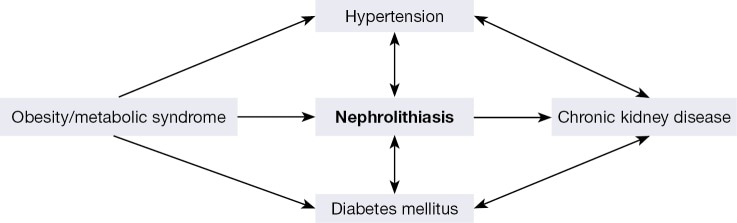
Existing theories suggest obesity, metabolic syndrome, or diabetes as precursors of kidney stone formation (Figure 4). Such alterations are also associated with chronic kidney disease and kidney injury [De Vries 2014]. Since these pathologies are defined by a common intermediate clinical feature of endothelial dysfunction, they are frequently associated with inflammatory biomarkers such as reactive oxygen species, alongside upregulated Piezo1 and TRPV4 channel proteins [Diaz-Ricart 2020].
Reactive oxygen species and oxidative stress are associated with kidney diseases to trigger endothelial inflammation and recruit inflammatory cells, while inducing the osteogenic nature of renal epithelial cells for calcification [Gambaro 2004]. Oxidative stress can affect some kidney structures including the glomeruli, tubules, and renal blood vessels to propagate inflammation, leading to kidney fibrosis and impaired functionalities – as observed at the renal papillary tip region of stone forming kidney patients [Khan 2012, Saenz-Medina 2022].
Reactive oxygen species (ROS) are produced by both mitochondria and NADPH oxidase as a major source of ROS in the kidney, in the presence of Angiotensin II. Mechanobiology brings together a few oxidative stress-related pathways to regulate cell injury and cell death (Figure 5). For example, renal epithelial cell injury triggered by hyperoxaluria is mediated by the accompanying oxidative stress and endothelial dysfunction [Drobnik 2024, Khan 2014]. And co-incubations of both endothelial and renal proximal tubule epithelial cell types administered with potent antioxidants can protect against epithelial tubular injury by hyperoxaluria, to decrease oxidative stress in tissues, and restore tissue function [Sarica 2012].
In a nutshell, the mechanosensors of Piezo1 and TRPV4 activate during high shear and pressure-induced mechanotransduction to increase intracellular calcium and trigger endothelial dysfunction and fibrosis. These ion channels also react to increasing microenvironmental stiffness that accompany calculi formation – in a positive feedback loop (Figure 5b).
Both caspase and calpain cascades are tightly interrelated at the boundaries of apoptotic and necrotic cell death during pathological biomineralization. Similarly, biomarkers of intracellular oxidative stress lend a primary mechanism to induce tissue injury, and facilitate hypoxia, apoptosis and necrosis-driven renal calcification (Figure 5) [Drobnik 2024, Saenz-Medina 2022]. These experiments reiterate our hypothesis of the activation of a potential redox regulatory biological switch at the renal papillary tip, at the onset of pathological fluid shear and oxidative stress.
Mechanisms of kidney stone formation and some fundamental experiments in animal models
Technically, the mechanism of human kidney stone formation can be classified into four categories -
- Growth over Randall’s plaque, as seen in idiopathic calcium oxalate (CaOx) stone formers, where most calculi are formed on Randall’s plaque.
- Growth across Bellini duct plugs seen mostly in calcium oxalate and calcium phosphate formers.
- And microlithiasis formation apparent in the inner medullary collecting ducts observed during cystinuria, and
- The formation in free solution within renal calyces or the renal collecting system, observed in calcium oxalate stones in patients with hyperoxaluria, and brushite or hydroxyapatite stone formers that constitute the core of Randall’s plaque (Rao 2019, Evan 2014).
Oxidative stress affects the first two mechanisms via endothelial injury during stone formation [Saenz-Medina 2022]. In a more general mechanism, calcium phosphate builds an initial deposit on the basement membrane of the loop-of-Henle as a nidus for subsequent calcium stone formation. Accompanying renal injury increases the likelihood of crystal attachment to renal epithelial cells, and injured cells can promote a stone nucleus for crystal aggregation to form a kidney stone. In oxalate associated injury of renal epithelial cells, both crystal-cell interaction and apoptosis form the basis of calcium oxalate stones and is accompanied with a loss of plasma membrane integrity, necrotic changes, and apoptosis [Khan 1999].
Renal epithelial cells exposed to oxalate, calcium oxalate or calcium phosphate crystals can rapidly bind to the surface of epithelial cells for crystal endocytosis. Subsequently increasing the expression of specific encoding genes such as transcription factors, matrix regulators and inflammatory markers as well as anti-inflammatory molecules including osteopontin, osteonectin, and fibronectin to induce inflammation, extracellular matrix production, and fibrosis to modulate biomineralization (Figure 5) [Khan 2004]. A list of urinary macromolecules produced during crystallization and inflammation is outlined on Table 1 [Khan 2014, Khan 2021].
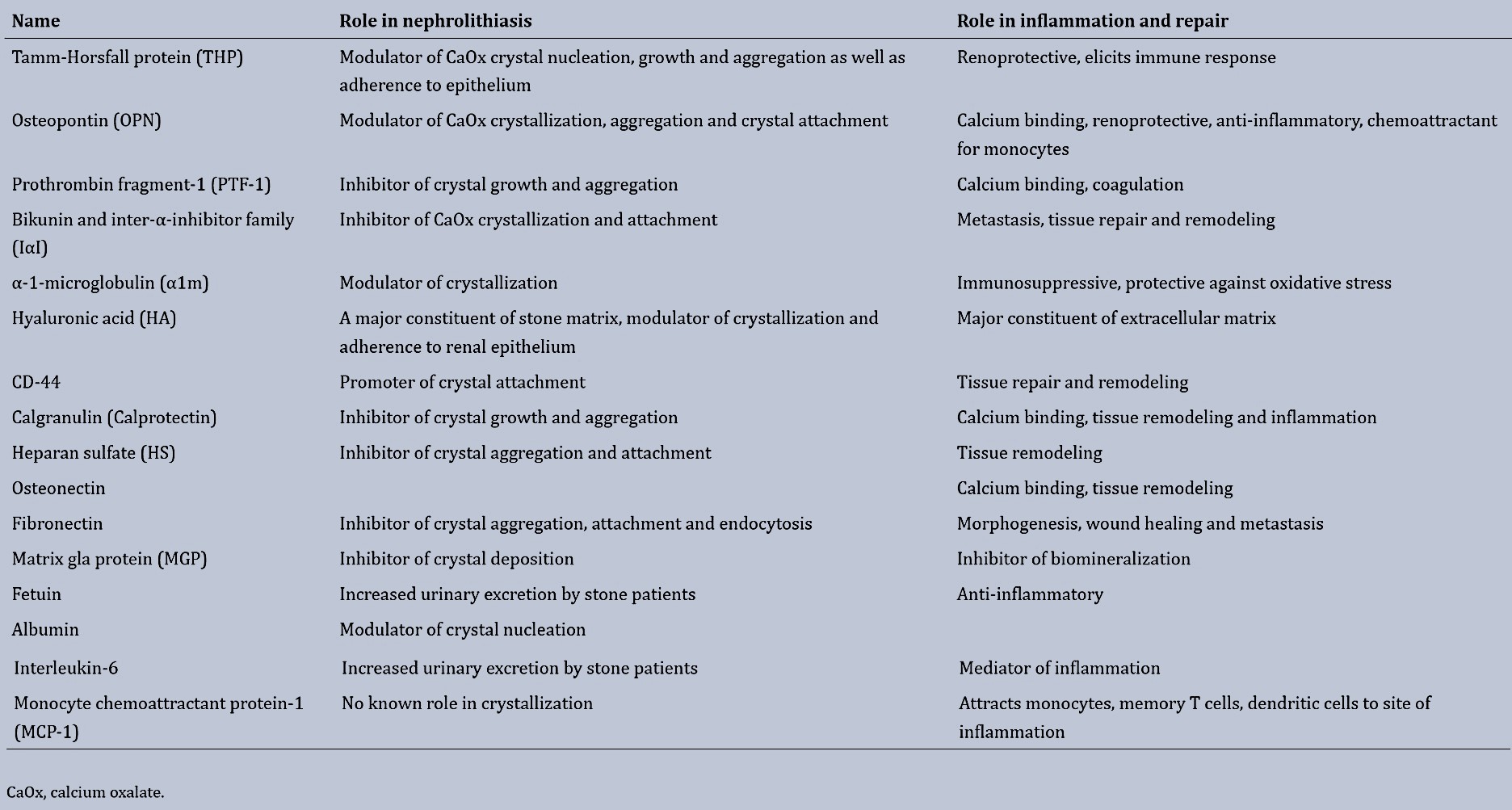
Table 1: Urinary macromolecules and their role in crystallization and inflammation [Khan 2014].
In an interesting turn of events, when exposed to calcium oxalate crystals, renal cells secrete superoxide and antioxidants as free radical scavengers to ameliorate cellular injury. Mitochondria is the site of antioxidant secretion and the most common source of reactive oxygen species generated as byproducts of the mitochondrial respiratory chain [Khan 2014]. The idea of an oxidative stress-mediated inflammatory response giving way to the deposition of calcium oxalate crystals was proven in animal models of hyperoxaluria [Thamilselvan 1997].
Pharmaceutically, inhibiting the renin-angiotensin aldosterone system via doses of spironolactone and atorvastatin is a well explored key pathological pathway to attenuate kidney disease in rodent models [Ruster 2006, Ng 2011, Jeewandara 2015] in order to circumvent hypertension associated shear-flow, and block the deposition of calcium oxalate crystals in rat kidneys of hyperoxaluria [Umekawa 2004, Tsujihata 2011].
A series of functional assays – pathological mechanotransduction on a kidney-on-a-chip
Given the pivotal role of the endothelium during physiologic and pathological outcomes, several functional assays can be carried out with endothelial cells, to inhibit piezo channels, and protect against shear-stress induced actin disorganization during renal fibrosis and calcification [Swain 2021]. These experiments can determine the impact of TRPV4 activation that mediates Piezo1-driven F-actin and adherens junction disruption. Shear-force studies can assess the combined role of TRPV4 and Piezo1 during sustained calcium elevation using human umbilical vein endothelial cells (HUVECs) [Schulz 2002].
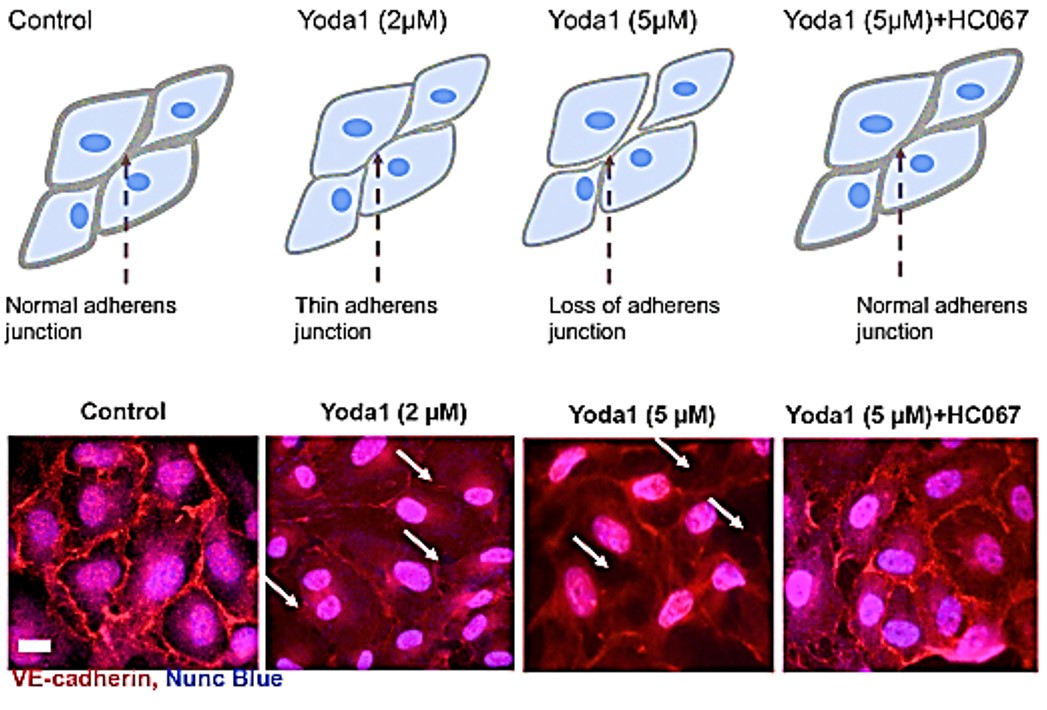
Figure 6: The TRPV4 antagonist HC067 prevents the Piezo1-mediated loss of adherens junctions in human umbilical vein endothelial cells. The accumulation of the adherens junction protein VE-cadherin at HUVEC junctions in monolayer cultures is shown by red immunostaining along the membranes of endothelial cells. The application of the Piezo1 agonist Yoda1 for 30 minutes caused thinning or loss of VE-cadherin in endothelial cells, while the HC067 antagonist of TRPV4 prevented the Yoda1-induced loss of VE-cadherin. Credit: [Swain 2021].
A series of functional assays that highlight pathological flow-mediated mechanotransduction can be carried out using HUVECs, human embryonic kidney 293 (HEK293T) cells, and human primary kidney glomerular endothelial cells plated as 3-D cultures on a kidney-on-a-chip platform to emulate the pathological microenvironment of the human kidney papillary tip region at the onset of endothelial dysfunction and renal calcification [Kaarj 2019].
Functional assays have already shown that administering the Piezo1 agonist Yoda1 caused cell retraction to produce paracellular gaps at adherens junctions via cell surface area reduction, through beautifully rendered experimental detail (movie 1) (Figure 6) [Swain 2021]. Administering the TRPV4 blocker HC067 can prevent cellular disruption caused by the Piezo1 agonist Yoda1, to confirm the combined pathological associations of the two mechanosensory ion channels in vivo.

Movie 1: A high concentration of the Piezo1 agonist Yoda1 caused cell retraction and produced paracellular gaps in monolayer cultures due to the reduction in the cell surface area. Cellular disruption due to Yoda1 was absent after adding the TRPV4 blocker HC067 into the media. Credit: [Swain 2021].
Loss-of-function assays can inhibit the shear-stress induced elevation of calcium ions by applying the Piezo1 antagonist GsMTx4. Treatment with a cytoplasmic and secretory blocker of phospholipase A2 (PLA2) too can inhibit calcium ions, to regulate Piezo1 [Khan 2004]. Flow-mediated endothelial mechanotransduction can be easily emulated on a kidney-on-a-chip instrument by introducing pathological fluid-shear stress, to mimic disease etiology via varied mechanisms (Figure 7).
Co-cultures of renal endothelial cells and renal proximal tubule epithelial cells provide insight to mechanoreceptor activation via inflammatory biomarker upregulation. Such insights can protect against the resulting epithelial tubular injury (typically observed with hyperoxaluria), by administering antioxidants to restore endothelial function [Sarica 2012].
Activating both Piezo1 and TRPV4 results in an initial increase of calcium, followed by a sustained elevation of calcium ions. Meanwhile, high and prolonged shear stress can be blocked by inhibiting the TRPV4 channel opening alone, to show the interlinked nature of the two mechanosensory ion channels [Saenz-Medina 2022], [Swain 2021].
Since basic science experiments already provide evidence to the necessity of both ion channels for shear-stress mediated F-actin disorientation [Swain 2021]. Microfluidic platforms are ideal to carryout shear flow-mediated experiments with human cells, to identify F-actin dysregulation and observe the concerted intracellular effects of Piezo1 and TRPV4. Additionally, aquaporins are another family of membrane proteins that function as water-permeable channels whose impairment can contribute to nephrogenic diabetes. Their role during hypercalciuria and renal concentrating defects leading to renal calcification can be further determined with histology and functional studies for further therapeutic investigations on a chip [Noda 2010, Procino 2012].
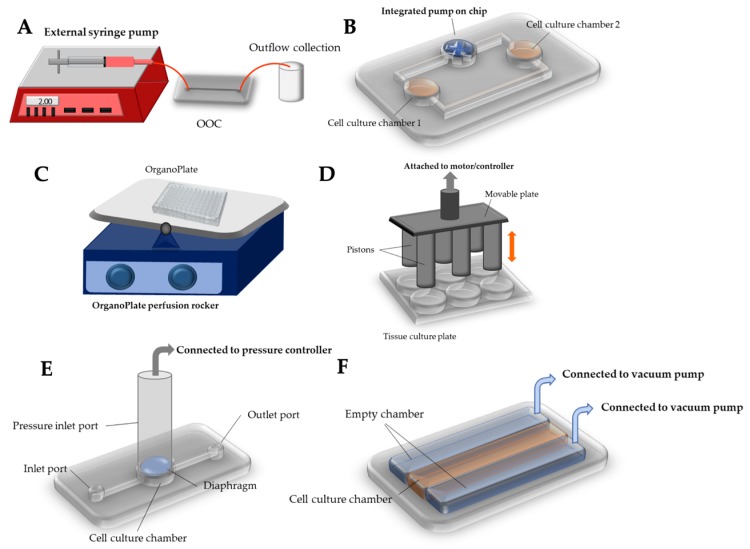
Figure 7: Innovative methods and equipment in use to deliver pathological mechanical stimuli to the organ-on-a-chip system aside from regular shear flow. A) external syringe pump to deliver shear flow, B) integrated micropump within a microfluidic organ-on-a-chip system to drive either laminar or pulsatile flow, C) compressive force delivered on a tissue culture plate, E) compressive force delivered by a pressure regulator, F) stretch or strain generated by periodically applying vacuum to the peripheral chambers to allow the main chamber to stretch cyclically [Kaarj 2019].
Functional assays – emulating oxidative stress on a chip
Recreating oxidative stress on an organ-chip instrument deals with the fact that reactive oxygen species are key regulators of disease and unite many mechanisms for calcium oxalate stone formation [Khan 2014]. Tissue culture studies conducted with renal epithelial cells exposed to high concentrations of oxalate, calcium oxalate or calcium phosphate crystals have caused injury to cell types [Khan 2014, Koul 1994].
Such cell exposure led to the secretion of superoxide in an extracellular oxidative burst. The resulting cellular injury can be ameliorated with antioxidants and free radical scavengers such as catalase and superoxide dismutase to protect cells from oxalate induced injury [Gaspar 2010, Thamilselvan 2000]. Additionally, the interaction of calcium oxalate monohydrate crystals with renal cells can cause altered gene expression, DNA synthesis and cell death, factors mediated by the p38 MAPK signaling pathway (Figure 5) [Koul 2002, Hammes 1995].
In animal models of calcium oxalate nephrolithiasis and hyperoxaluria, the treatment with vitamin E improved tissue levels of antioxidant enzymes, reduced injury and eliminated calcium oxalate crystal deposition in the kidneys [Thamilselvan 2005]. The administration of an antioxidant apocynin nearly completely reversed the effects of hyperoxaluria in animal models and reduced the deposition of calcium oxalate crystals in kidneys as a proof-of-concept (Figure 8) [Khan 2014]. Functional assays of this nature can be replicated with clinical samples of epithelial and endothelial cell types obtained from renal stone formers, with exposure to calcium oxalate crystals on a microfluidic platform alongside pathological shear, to observe therapeutic effects of small molecule antioxidant drugs.
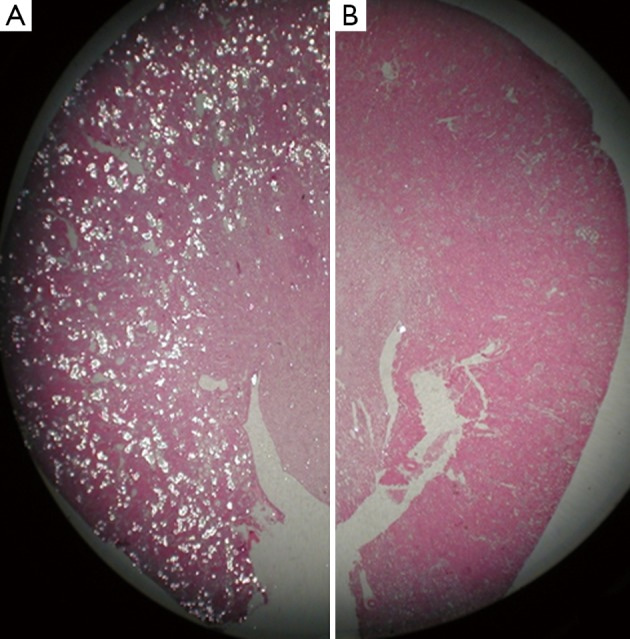
Figure 8: Experimental outcomes of hematoxylin and eosin-stained paraffin sections of a kidney obtained from a hyperoxaluric rat model. Tissue sections were examined under light microscopy to indicate; A) a hyperoxaluric rat with renal tubules full of birefringent calcium oxalate crystals, b) hyperoxaluric rat receiving an antioxidant apocynin showed only the presence of a few birefringent crystals of calcium oxalate. Credit: [Khan 2014].
Is there a biological switch at the renal tip that triggers the onset of renal calcification?
The range of basic science studies provide a foundation for bioinspired engineering, to investigate renal calcification on a chip. I conceptualized, designed, and carried out the following first-in-study experiments in our project of bioengineering and mechanobiology, completed at the University of California, San Francisco, to arrive at the described outcomes (detailed here is a snippet of the whole).
The hypothetical presence of a biological switch that can turn on/off at the onset of hypoxia in the renal papillary tip region of stone formers is a new concept that can be determined at the biomolecular level with first-principles investigations. For example, the hypoxia inducible factor 1α (HIF1α) is associated with progressive oxidative stress-related chronic renal injuries, tissue fibrosis and genetic expressions of vimentin – a biomarker of epithelial-to-mesenchymal transitions [Higgins 2007]. The expression of vimentin and other pathological biomarkers of fibrosis such as collagen and calcium deposits were detected with histology studies of the renal tip of clinical stone-former patient samples, to subsequently inform the development of functional assays on a chip, to investigate similar biomarkers of interest (Figure 9) [ Jeewandara 2023, ref 47].
Using histology sections for analyses with an artificial neural network derived from quantitative pathology (quPath) software, I first measured the key biomarkers of renal calcification, and these outcomes guided the subsequent development of organ-on-a-chip functional assays. In detail, the epithelial-to-mesenchymal transitions visualized with vimentin-DAB are a precursor of fibrosis and subsequent calcification, the biomarker was significantly expressed in renal stone forming patient samples (SF) compared to non-stone formers (NSF). Collagen is another biomarker of renal injury and fibrosis that we visualized with Masson's trichrome for quantification, upregulated in the renal papillary region due to shear stress. Stone forming patients showed significantly higher levels of collagen deposition vs. non-stone formers. We used the Alizarin red dye to visualize and identify renal calcification, where calcium deposits appeared as crimson aggregates across the tissue. As expected, renal stone-formers showed higher levels of calcium deposits due to dye uptake compared to NSF. In our study, the papillary tip region of all stone-forming patient samples exhibited the key biomarkers of collagen deposition (Masson's Trichrome), epithelial-mesenchymal transitions (vimentin-DAB), and calcification (Alizarin Red) in greater quantity than non-stone formers (Figure 9) [Jeewandara 2023].
The M1 pro and M2 anti-inflammatory macrophage cell lines used in the functional assays were then differentiated in a stepwise process, first by using stone forming patient-derived whole blood cells, followed by lymphocyte extraction to generate peripheral blood mononuclear cells as precursors of macrophage differentiation.
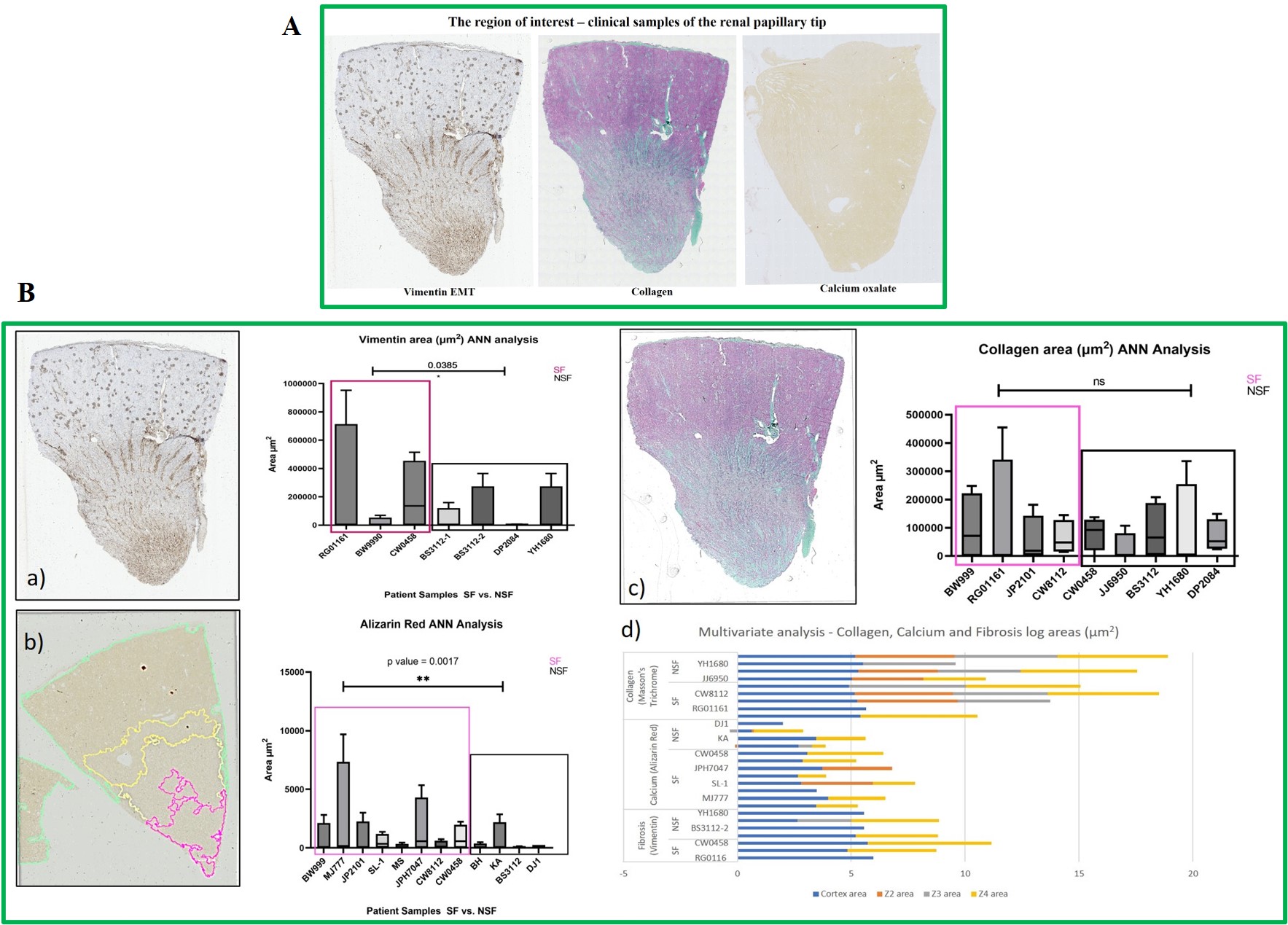
Figure 9: The region of interest: the renal papillary tip A) The human renal papillary tip region (from L-to-R) stained with DAB-Vimentin antibody to demonstrate epithelial-to-mesenchymal transitions during renal fibrosis, followed by trichrome stains to indicate collagen deposition and renal fibrosis, and alizarin red to identify calcium deposits (red). B) Histology-stained renal papillary tips were quantified with an artificial neural network by using quPath software to measure regions of interest a) vimentin for endothelial-mesenchymal transitions, b) the ANN-driven segmentation method represented with Alizarin red dye during renal calcification and quantification, and c) Masson’s trichrome stained collagen area, d) log values of the multivariate analyses of all samples across the renal papillary cross-sections. Image credit: Author’s own.
The ‘hypoxia response via HIF activation’ pathway is often upregulated in the transcriptome of stone forming patients and is a key mechanism of HIF1α stabilization [Han 2013]. Prolyl hydroxylase domain 2 (PHD2) protein is a primary oxygen regulator and a site-specific zinc finger enzyme that can hydroxylate HIF1α and degrade it under normoxia [Arsenault 2016]. Under hypoxia, however, this process of degradation does not occur, leading to the stabilization of HIF1α instead, and its subsequent activation as a pathological hallmark of kidney injury and stone formation (Figure 10) [Meneses 2016, Arsenault 2016].
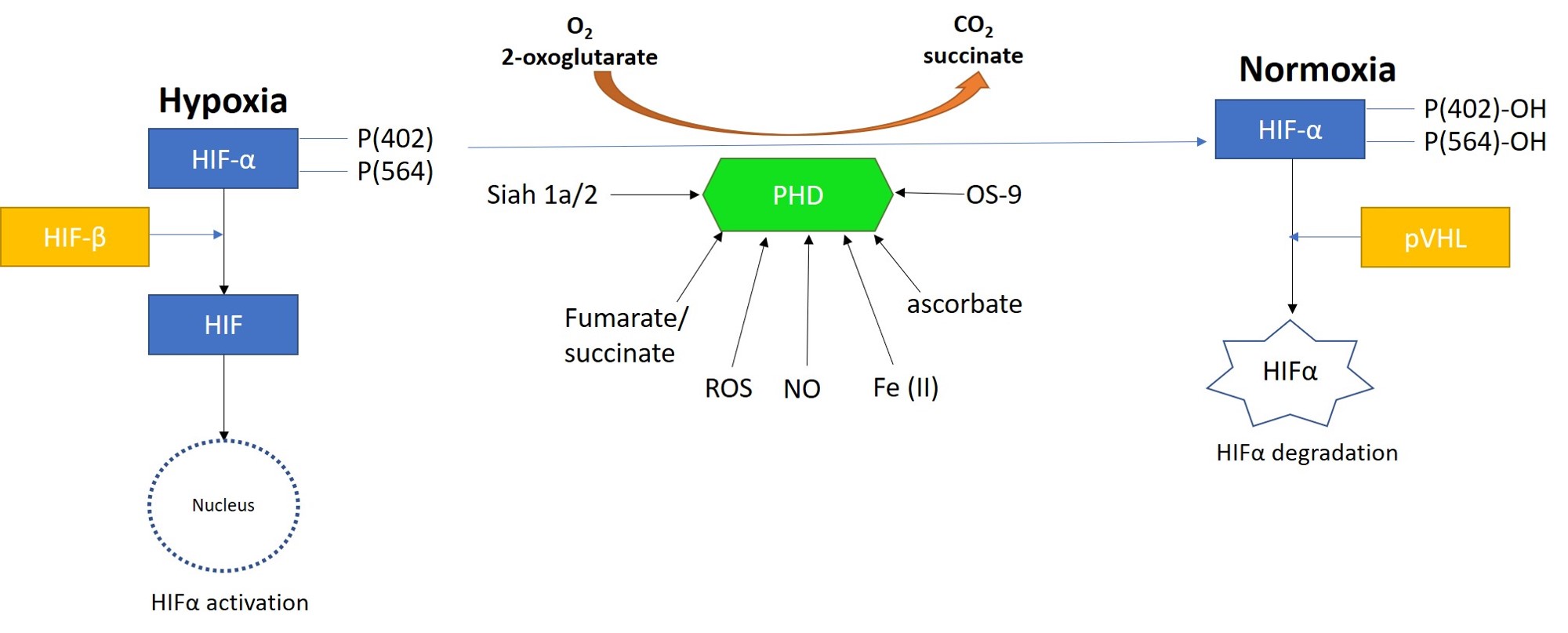
Figure 10: Prolyl hydroxylase domain 2 (PHD2) is a key oxygen sensor in mammals that post-translationally modifies hypoxia-inducible factor α (HIF1α) to target it for degradation by forming a complex with the Von Hippel-Lindau factor (pVHL). Reactive oxygen species, nitrous oxide, osteosarcoma amplified 9, ascorbate, succinate, iron, and protein ligase components can regulate the function of PHDs. Figure credit: Author’s own.
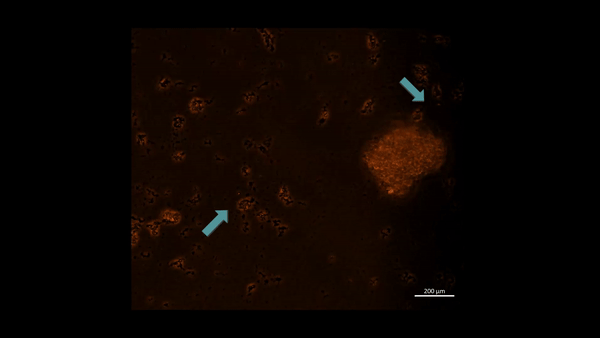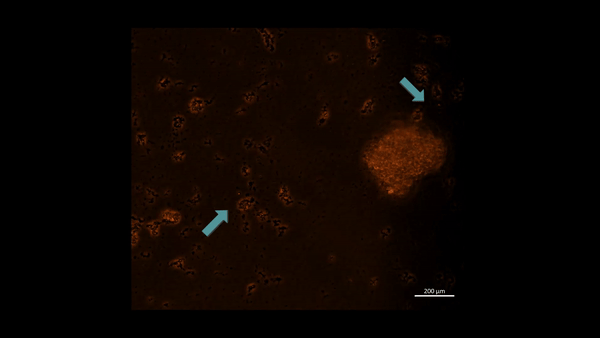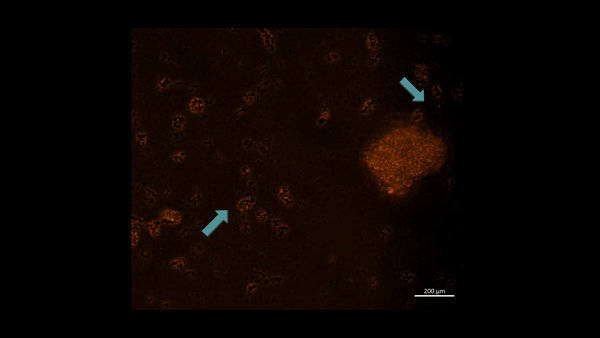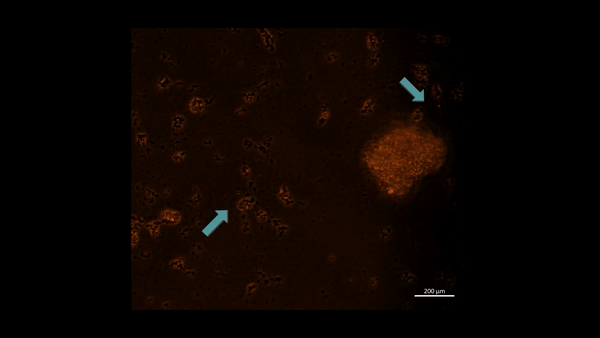
First-in-study outcomes have already verified the upregulation of PHD2 biomarkers during oxidative stress induced cell death via in vitro apoptotic assays, using renal patient-derived M1 pro and M2 anti-inflammatory macrophage cell lines cultured on a microfluidic kidney-on-a-chip instrument and on a culture plate, separately (Movie 2ab) [Jeewandara 2023]. These early outcomes of oxidative-stress induced immunofluorescence highlighted the biological switch-like activation of the PHD2 enzyme during hypoxia, to regulate the HIF1α pathway of stone-forming patients, with relevance at the renal papillary tip regions in vivo.
Since chronic hypoxia response and an apoptosis-driven oxidative stress signal underly the progression of several renal stone forming pathways [Saenz-Medina 2022], the experimental capacity to reveal a switch or regulator of pathological oxidative stress – for eventual therapeutic intervention is a significant observation, with scope for applications in the clinical treatment of nephrolithiasis.

Movie 2a: Normoxia-on-a-chip/plate (fluorescence imaging): Miniature motile cluster-forming M2 anti-inflammatory macrophages trawl through the top chamber of the organ-on-a-chip instrument by day 3 of cell culture. Under normoxia the highly confluent and motile cells are active, although activated PHD2 immunofluorescence is not observed predominantly among the immunofluorescence-stained negative control cells at normoxia. Image credit: Author’s own.
An innate defense mechanism –regulating M1 and M2 macrophages during nephrolithiasis
The evidence of the origin of renal disease thus far is insufficient to facilitate new curative therapies to attenuate kidney stone formation [Taguchi 2021]. Renal and peripheral macrophages can be involved during inflammation for renal crystal development suppression, as a potential next-generation avenue of immunotherapy. Technically, lithogenic environments of calcification can trigger inflammation, and upregulate reactive oxygen species for renal cell impairment and M1 macrophage-facilitated calcium oxalate stone deposition (Figure 11) [Khan 2016, Khan 2021].
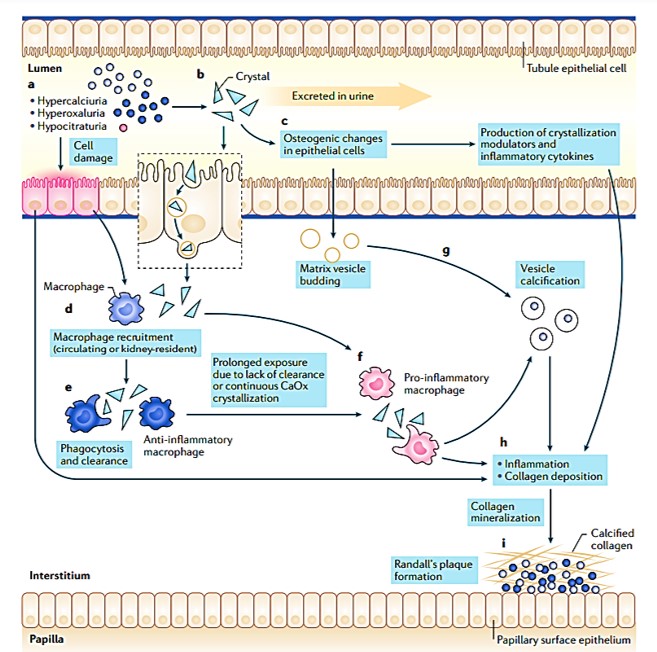
Figure 11: A proposed model for the formation of Randall’s plaque. Steps a-h recap the risk factors of hyperoxaluria, hypercalciuria, and hypocitraturia and other factors involved in the pathogenesis of nephrolithiasis to cause epithelial damage, the production of calcifying vesicles, increased pro-inflammatory macrophages vs. anti-inflammatory macrophages, collagen deposition, mineralization, and Randall’s plaque formation [Khan 2021].
Autophagy provides a primary defense mechanism against cellular impairment via endocytosis [Taguchi 2021] where macrophages form an innate defense mechanism, in order to clear crystal deposits from renal tissues (Figure 11). First reported in 1999, this avenue can be explored to offer a solution to develop treatments for kidney stones [de Water 1999]. To accomplish this, it is necessary to understand the role of macrophages during renal crystal formation as they migrate to calculi deposition sites to engulf the crystals [Murray 2014].
First-in-study outcomes have already shown an initial promise of differentiating macrophages to form M1 pro- and M2 anti-inflammatory cell types (Figure 12). Whole blood cells derived from stone-forming patients were differentiated via precursors of lymphocytes and peripheral blood mononuclear cell types to generate the macrophage cell types in vitro (Movie 3ab) [Jeewandara 2023].
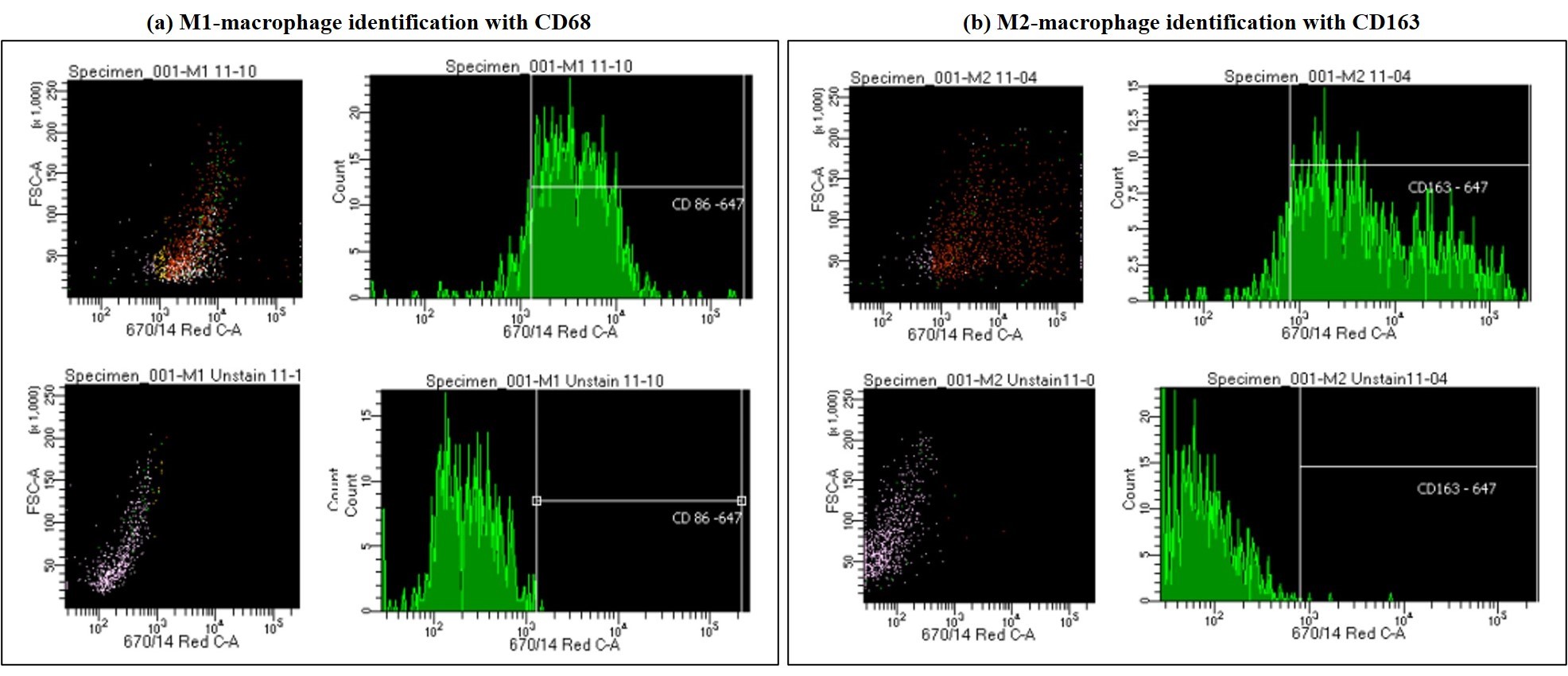
Figure 12: Using fluorescence-activated cell sorting to a) distinguish M1 polarized macrophages stained with CD86 vs. the unstained M1 polarized macrophages, and b) distinguishing M2 polarized macrophages stained with CD163 vs. the unstained M2 polarized macrophages. Image credit: Author’s own.
The study outcomes demonstrated the versatility of organ-chip instruments to support the proliferation of the miniature, cluster-forming motile M1 and M2 macrophages, when compared to those macrophage aggregates formed in clusters on the tissue culture plate surface. Macrophages differentiated in the lab were identified by fluorescence-activated cell sorting to distinguish between the two types of polarizations (Figure 12). The M2-like macrophages have greater capacity to phagocytose crystals than M1-like macrophages that are instead involved in the process of crystal deposition, therefore the capacity to regulate M2 polarization has therapeutic value [Taguchi 2021].

Bioinspired engineering on a chip to investigate renal calcification – basic science is not just a foundation.
To wrap things up, all complex diseases result from a concerted effort of several dysfunctional signaling pathways that come together to establish disorder – the pathology of calcification follows a similar trajectory [Khan 2021]. Bulk RNA-sequencing data acquired from renal papillary tip regions of stone forming patients have already revealed the upregulation of several pathological mechanisms during renal calculi formation [Jeewandara 2023].
These gene analyses include the dysregulation of the actin cytoskeleton, upregulation of phagosomes, hypoxia response via HIF activation, apoptosis signaling pathways, p38 MAPK pathway activation and oxidative stress [Koul 2002]. In agreement with transcriptomics, first-principle based functional assays in basic science have indicated the presence of pathological biomarkers of oxidative stress, F-actin disruption, and macrophage infiltration as corresponding biomechanisms at calculi forming regions of the renal papillary tip.
Preliminary experimental outcomes highlighted the biological switch-like activation of prolyl hydroxylase2 (PHD2) enzyme to regulate the hypoxia-inducible factor 1α (HIF1α) and activate a route of oxidative stress in the renal papillae of stone-forming patients. First-in-study outcomes showed the presence of PHD2 in the form of a pathological switch that acts to regulate HIFα in the renal papillary region at the onset of hypoxia, to attenuate related endothelial dysfunction, renal injury, and calcification. This oxidative stress pathway can be further explored with apoptotic assays and redox biomarkers to observe hypoxia on a chip and a plate. The outcomes can inform the development of small molecule antioxidants for therapeutic intervention, to attenuate the origin of pathological biomineralization (Figure 13).

The histology work also delineated the significantly upregulated expression of pathological biomarkers of renal fibrosis, epithelial-mesenchymal transitions, and subsequent calcification represented via collagen deposits, vimentin, and alizarin red dye deposits, respectively, in stone forming patients to be greater than in non-stone forming patients. The results presented herein are only a snippet of the completed mechanobiology and biomolecular project to emulate biomarkers of renal calcification; through bottom-up engineering a pathological cascade on an organ-chip instrument. Future articles will include further iterations of the completed research works.
Similarly, the concerted efforts of shear-force induced Piezo1, TRPV4 and ROS functions that contribute to cytoskeleton disruption at the onset of tissue injury and calcification can be emulated on a microphysiological environment, while macrophages that provide an innate defense mechanism by engulfing calcium deposits or by promoting calculi deposition can be investigated to promote new therapeutics accordingly. This research focus on basic science reiterates the idea that while the redeeming qualities of basic biology may not become immediately apparent, the expansion of new knowledge has often led to breakthroughs in medicine, years and decades later. The pure research field coupled with bioengineering is a way of thinking to find answers.
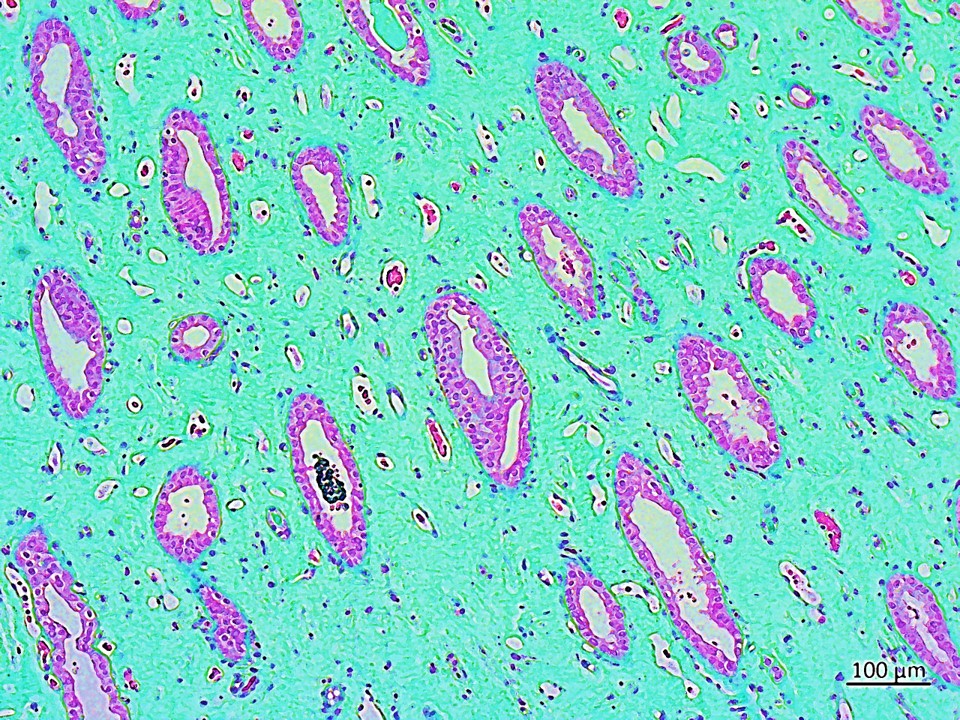
Header Image: A region of a clinical sample of the renal papillae of a stone-forming patient, stained with trichrome to indicate pathological collagen deposition to represent interstitial fibrosis in the renal papillary tissue (20 x Magnification). Image credit: Author's own.
References
- Saenz-Medina J. et al. Endothelial dysfunction: an intermediate clinical feature between urolithiasis and cardiovascular diseases, MDPI, 2022.
- Aydin H. et al. Ethylene glycol induced hyperoxaluria increases plasma and renal tissue asymmetrical dimethylarginine in rats: a new pathogenetic link in hyperoxaluria induced disorders, Journal of Urology, 2010.
- Saenz-Medina J. et al. Urolithiasis develops endothelial dysfunction as a clinical feature, Antioxidants (Basel), 2021.
- He B. et al. Myeloid Piezo1 deletion protects renal fibrosis by restraining macrophage infiltration and activation, Hypertension, 2022.
- Valverde M. et al. Biomimetic models of the glomerulus, Nature Reviews Nephrology, 2022.
- Coste B. et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels, Science, 2010.
- Ranade S. et al. Mechanically Activated Ion Channels, Neuron, 2015.
- Cheng Y. et al. TRPV1 and Piezo: the 2021 Nobel Prize in Physiology or Medicine, IUCrJ 2022.
- Deng Z. et al. Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms, Nature Structural and Molecular Biology, 2018.
- Swain S. et al. Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress, Journal of Biological Chemistry, 2021.
- Dalal P. et al. Endothelial Cell Calcium Signaling during Barrier Function and Inflammation, American Journal of Pathology, 2020.
- Yoneda M. et al. PIEZO1 and TRPV4, which are distinct mechano-sensors in the osteoblastic MC3T3-E1 cells, modify cell-proliferation, MDPI 2019
- Mochizuki T. et al. The TRPV4 cation channel mediates stretch-evoked Ca2+ influx and ATP release in primary urothelial cell cultures, Journal of Biological Chemistry, 2009.
- Strotmann R. et al. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity, Nature Cell Biology, 2000.
- Mendoza S. et al. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress, American Journal of Physiology, 2010.
- Li W. et al. TRPV4 inhibitor HC067047 produces antidepressant-like effect in LPS-induced depression mouse model, Neuropharmacology, 2021.
- Everaerts W. et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis, PNAS, 2010.
- Gambaro G. et al. Crystals, Randall's plaques, and renal stones: do bone and atherosclerosis teach us something? Journal of Nephrology, 2004.
- Duffield J. et al. Cellular and molecular mechanisms in kidney fibrosis, The Journal of Clinical Investigation, 2014.
- De Vries A et al. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease, The Lancet 2014.
- Diaz-Ricart M. et al. Endothelial Damage, Inflammation, and Immunity in Chronic Kidney Disease, MDPI, 2020.
- Khan S. et al. Reactive oxygen species, inflammation and calcium oxalate nephrolithiasis, Translational Andrology and Urology, 2014.
- Khan S. et al. Association of Randall's Plaques with Collagen Fibers and Membrane Vesicles, Journal of Urology, 2012.
- Sarica K. et al. Human umbilical vein endothelial cells accelerate oxalate-induced apoptosis of human renal proximal tubule epithelial cells in co-culture system which is prevented by pyrrolidine dithiocarbamate, Urological Research, 2012.
- Drobnik M. et al. Mechanosensitive cation channel Piezo1 is involved in renal fibrosis induction, MDPI, 2024.
- Rao C. et al. Effects of physical properties of nano-sized hydroxyapatite crystals on cellular toxicity in renal epithelial cells, Materials Science and Engineering: C, 2019.
- Evan A. et al. Mechanisms of human kidney stone formation, Urolithiasis, 2014.
- Khan S. et al. Crystal-cell interaction and apoptosis in oxalate-associated injury of renal epithelial cells, Journal of American Society of Nephrology, 1999.
- Khan S. et al. Role of renal epithelial cells in the initiation of calcium oxalate stones, Experimental Nephrology, 2004.
- Thamilselvan S. et al. Lipid peroxidation in ethylene glycol induced hyperoxaluria and calcium oxalate nephrolithiasis, Journal of Urology, 1997.
- Ruster C. et al. Renin-angiotensin-aldosterone system and progression of renal disease, JASN, 2006.
- Ng K. et al. Aortic stiffness is associated with vascular calcification and remodeling in a chronic kidney disease rat model, Renal Physiology, 2011.
- Jeewandara T. et al. Protective cardiorenal effects of spironolactone in a rodent model of polycystic kidney disease, CEPP, 2015.
- Umekawa T. et al. Effect of angiotensin II receptor blockage on osteopontin expression and calcium oxalate crystal deposition in rat kidneys, JASN, 2004.
- Tsujihata M. et al. Why does atorvastatin inhibit renal crystal retention? Urological Research, 2011.
- Schulz E. et al. Functional and biochemical analysis of endothelial (dys)function and NO/cGMP signaling in human blood vessels with and without nitroglycerin pretreatment, Circulation, 2002.
- Kaarj K. et al. Methods of Delivering Mechanical Stimuli to Organ-on-a-Chip, MDPI, 2019.
- Noda Y. et al. Aquaporins in kidney pathophysiology, Nature Reviews Nephrology,
- Procino G. et al. Calcium-sensing receptor and aquaporin 2 interplay in hypercalciuria-associated renal concentrating defect in humans. An in vivo and in vitro study, PLoS One, 2012.
- Koul H. et al. Oxalate-induced initiation of DNA synthesis in LLC-PK1 cells, a line of renal epithelial cells, Biochemical and biophysical research communications,1994.
- Gaspar S. et al. Effect of calcium oxalate on renal cells as revealed by real-time measurement of extracellular oxidative burst, Biosensors and Bioelectronics, 2010.
- Thamilselvan S. et al. Free radical scavengers, catalase and superoxide dismutase provide protection from oxalate-associated injury to LLC-PK1 and MDCK cells, Journal of Urology, 2000.
- Koul H. et al. COM crystals activate the p38 mitogen-activated protein kinase signal transduction pathway in renal epithelial cells, Journal of Biological Chemistry, 2002.
- Hammes M. et al. Calcium oxalate monohydrate crystals stimulate gene expression in renal epithelial cells, Kidney International, 1995.
- Thamilselvan S. et al. Vitamin E therapy prevents hyperoxaluria-induced calcium oxalate crystal deposition in the kidney by improving renal tissue antioxidant status, BJU International, 2005.
- Higgins D. et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition, Journal of Clinical Investigations, 2007.
- Jeewandara T. et al. The dynamics of pathological biomineralization in the renal papillae, World Congress of Nephrology 2023, abstract number: WCN23:0323, 2023.
- Han W. et al. Hypoxia-inducible factor prolyl-hydroxylase-2 mediates transforming growth factor beta 1-induced epithelial–mesenchymal transition in renal tubular cells, Molecular Cell Research, 2013.
- Arsenault P. et al. The Zinc Finger of Prolyl Hydroxylase Domain Protein 2 Is Essential for Efficient Hydroxylation of Hypoxia-Inducible Factor α, Molecular and Cellular Biology, 2016.
- Meneses A. et al. PHD2: from hypoxia regulation to disease progression, Hypoxia, 2016.
- Taguchi K. et al. Macrophage function in calcium oxalate kidney stone formation: A systematic review of literature, Frontiers Immunology, 2021.
- Khan S. et al. Randall's plaque and calcium oxalate stone formation: role for immunity and inflammation, Nature Reviews Nephrology, 2021.
- Khan S. et al. Kidney stones, Nature Reviews Disease Primers, 2016.
- de Water R. et al. Calcium oxalate nephrolithiasis: effect of renal crystal deposition on the cellular composition of the renal interstitium, American Journal of Kidney Disease,1999.
- Murray P. et al. Macrophage activation and polarization: nomenclature and experimental guidelines, Immunity, 2014.
I am an interdisciplinary researcher with a strong commitment to bioengineering, biochemistry, organ-chips and molecular biology. As well as biomechanics and biomineralization in the broader context of medicine. I completed my PhD at the University of Sydney Australia in December 2016, and travel often, find me on Twitter and irl.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in