From Metagenomes to Environments
Published in Microbiology
The paper in Nature Microbiology is here: https://www.nature.com/articles/s41564-018-0124-8
The ability to generate, store, process, and replicate information is one the defining properties of living organisms. In the words of Andreas Wagner, “organisms live and die by the amount of information they acquire about their environments” [1]. As we all know, the genome sequence codes for genes that when expressed as proteins perform most of the functions that living organisms are known for. For example, they may help bacteria uptake nutrients from their environments, metabolize, create biomass, and replicate. Similarly, a metagenome sequence contains information about the growth preferences of all the micro-organisms found within it.
Microbial metabolism
is a process with many degrees of freedom, especially in a complex
environment. This makes it difficult to model, but luckily there are
also a great deal of constraints that reduce the degrees of freedom.
For example, we can identify many of the metabolic functions that are
encoded on the microbial DNA and the stoichiometric balance in the
chemical reactions between the metabolites. Modeling these
constraints can reveal a subset of achievable states that a community
could exhibit.
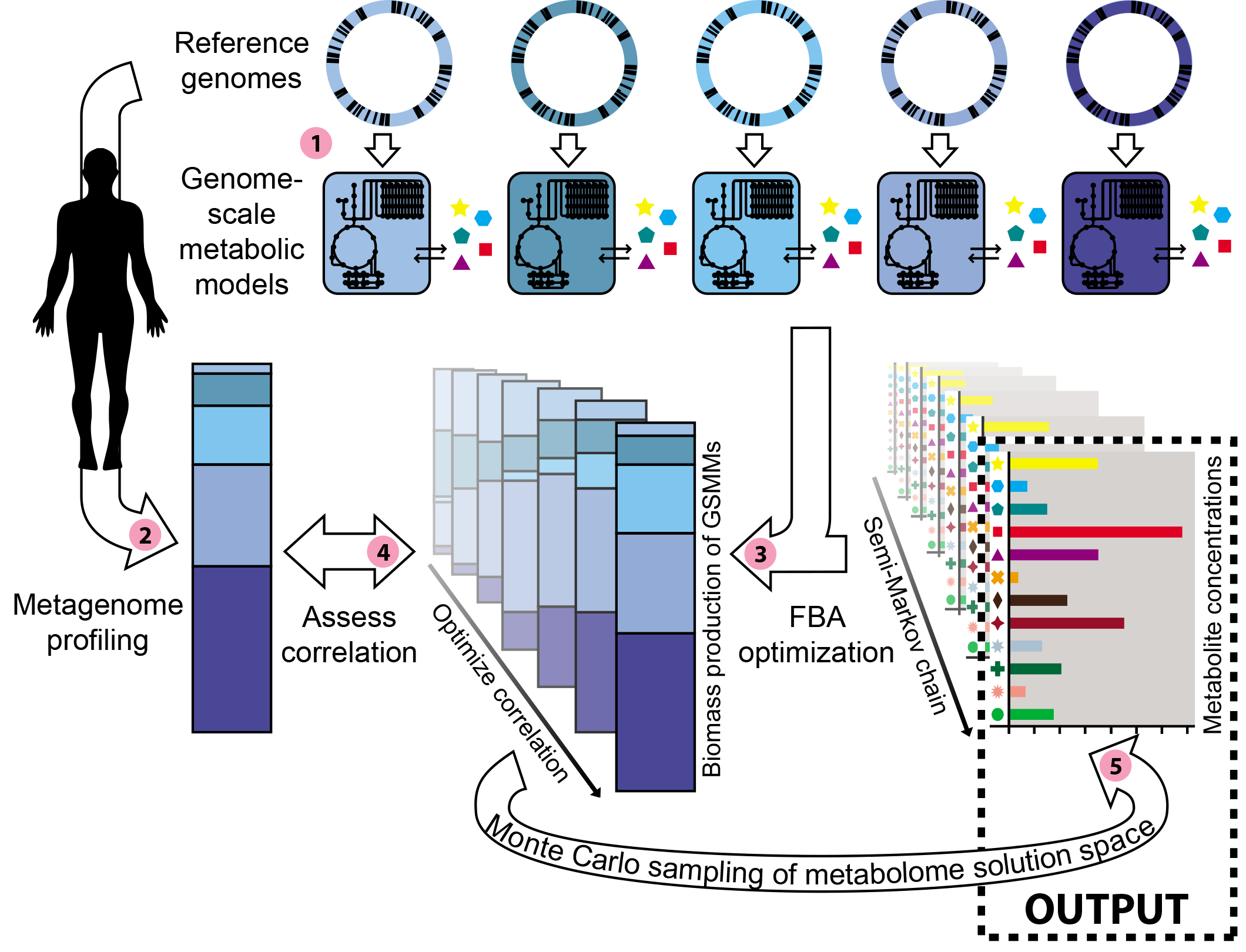
With these guiding
principles in mind, we decided to tackle a very fundamental question.
By analysing a metagenome (c.q. the relative abundances and functions
of the microbes found in a metagenomic dataset), can we predict which
metabolites were present in the environment where the sample was
taken? We wanted to answer this question with a modeling approach
that was unsupervised with respect to the environment and that
integrated the information about the metabolism and the relative
abundances of bacterial species. We implemented a bottom-up strategy
with no community-wide objective, where each modeled bacterium
optimally uses its genetically encoded functions to create new
biomass from the metabolites provided by its environment. However,
the search space for the metabolic environment is massive, each
metabolite effectively serving as another dimension that complicates
the search. Thus, we included a heuristic into the algorithm to
search this highly dimensional space effectively for the metabolic
environment that best explains the metagenomic data.

After implementing the model, we were excited that we could actually find consistent metabolic compositions for several major human body sites. You can read all about these results in our paper, but one of the most exciting result was to find by modeling that the skin was enriched for metabolites that are commonly found in cosmetics. In other words, by combining skin metagenomes with detailed genome annotations, we confirmed the enrichment for cosmetic ingredients on skin, confirming a recent high-throughput metabolomics study [2] by only using recycled data! We are excited about the prospect of using metabolic models to gain knowledge about microbial communities and their environments, as well as other secrets that remain hidden in metagenomic datasets.
[1] Wagner 2007 “From bit to it: how a complex metabolic network transforms information into living matter”, BMC Syst Biol. 1: 33.
[2] Bouslimani et al. 2015 “Molecular cartography of the human skin surface in 3D”, Proc Natl Acad Sci USA 112: E2120-9. doi: 10.1073/pnas.1424409112.
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in