Introducing scCross: a deep generative model for unifying single-cell multi-omics with seamless integration, cross-modal generation, and in-silico exploration
Published in Bioengineering & Biotechnology, Genetics & Genomics, and Mathematical & Computational Engineering Applications

The introduction of single-cell sequencing technology marks a new era in biological research, allowing scientists to analyze cellular heterogeneity with unprecedented detail. This advancement reveals complex cellular dynamics and has had significant impacts on fields such as cancer biology, neurobiology, and drug discovery. However, the data generated by these technologies are often highly complex and diverse, leading many existing computational tools to provide a limited perspective focused on specific data modalities. This limitation hinders a comprehensive understanding of the cellular landscape.
Challenges in Multi-Omics Data Integration and Generation
Integrating single-cell multi-omics data effectively remains a significant challenge in the field. Many existing methods depend on matched multi-omics datasets, which are often difficult to obtain, limiting the scope of analyses. These limitations result in insufficient integration of unmatched data and difficulties in managing noise and information loss. Even methods designed to handle multiple data modalities face persistent challenges, such as extracting common features across modalities and managing nonlinear transformations. The imbalance in the availability of different omics data types further complicates this issue; for instance, single-cell epigenomics data is often far less accessible compared to its transcriptomics counterparts. This scarcity not only hinders multi-omics analysis but also limits the potential for discovering comprehensive biological insights. These challenges highlight the need for more robust and flexible approaches to multi-omics data integration and generation, capable of overcoming the existing gaps and limitations in the field.
Developing Integrated Methods for Integration, Generation, Perturbation, and Downstream Analysis
To address these challenges, we propose scCross. This method excels in integrating single-cell multi-omics data and is particularly unique for its ability to generate cross-modal single-cell data. This capability bridges rich and scarce data modalities, allowing for a more comprehensive depiction of cellular states. Another key feature of scCross is its high-fidelity simulation of single-cell multi-omics data and support for computational perturbations. This enables virtual experiments of cellular interventions based on data integration, exploring potential strategies for cellular manipulation. By offering deep insights into cross-modal cellular dynamics, scCross not only enhances the utility of single-cell multi-omics research but also drives innovation and development in the field.
Integrating Multi-Omics Using Deep Generative Frameworks
The scCross model for integrating and generating single-cell multi-omics data leverages a deep generative framework that combines variational autoencoders (VAEs) and generative adversarial networks (GANs). This framework facilitates the seamless integration of single-cell multi-omics data, cross-modal data generation, multi-omics data simulation, and computational perturbations within and across modalities. The process begins by training VAEs for each modality to capture low-dimensional cell embeddings, enriched with gene set vectors for additional informational depth. These embeddings are then integrated into a common latent space, with a Jensen-Shannon (JS) divergence loss applied to minimize differences in data distributions across various omics. GANs are subsequently employed to fuse the modalities within this joint latent space. To further refine the integration, mutually nearest neighbor (MNN) cell pairs are used as anchors, guiding the alignment process and ensuring that embeddings of the same or similar cells across different modalities remain close in the joint latent space. This MNN-guided alignment results in a coordinated integration and distribution of modal data, ensuring robust and accurate multi-omics data integration.
Cross-Modal Generation Using Bidirectional Alignment
Beyond the integration of single-cell multi-omics data, the model also enables cross-modal single-cell data generation and perturbations. The bidirectional aligner is essential for this process, decoding shared latent embeddings into different modalities. Once trained, the model can generate single-cell data across modalities by encoding data from one modality into the latent space and then decoding it into another. Additionally, it simulates multi-omics data generation and performs computational perturbations both within and across modalities, uncovering potential regulatory changes in cellular states. By consolidating single-cell multi-omics data into a unified latent space and supporting cross-modal integration, scCross lays the foundation for a wide range of single-cell multi-omics applications, particularly in scenarios where certain omics data are limited or unavailable.
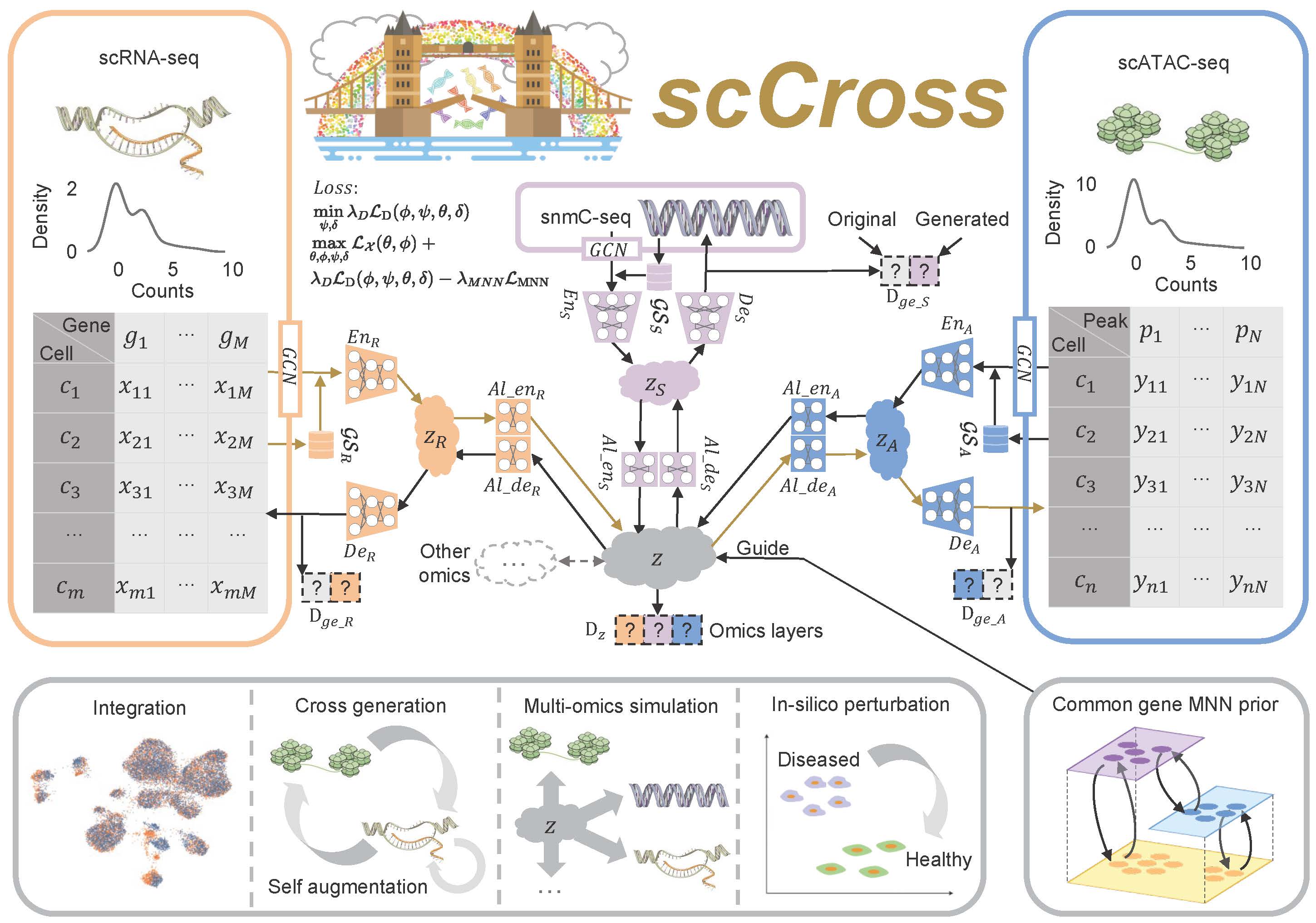
Fig. 1: Overview of the scCross method. scCross employs variational autoencoders for each modality to capture latent cell embeddings for different single-cell omics. During single-cell data integration, the method incorporates biological priors, such as gene set matrices, as additional features. It then uses additional variational autoencoders and a bidirectional aligner to merge these enriched embeddings into a shared latent space z. The bidirectional aligner is crucial for cross-modal generation, with brown arrows indicating the transition from scRNA-seq to scATAC-seq. Mutual nearest neighbor priors ensure alignment accuracy. A discriminator maintains integration across omics while ensuring the generated data’s completeness and consistency. scCross provides a robust toolkit for single-cell data integration, supporting cross-modal data generation, single-cell data enhancement, multi-omics simulation, and computational perturbations, offering great flexibility in addressing various single-cell multi-omics challenges.
Validation of scCross
We validated scCross across diverse datasets encompassing various single-cell omics. The results indicate that scCross performs effectively in single-cell multi-omics data integration, cross-modal generation, multi-modal simulation, and computational perturbation tasks, as confirmed by multiple metrics and downstream analyses. These findings suggest that scCross is a valuable tool for facilitating single-cell multi-omic explorations and enhancing data utilization, supporting researchers in gaining deeper insights into single-cell multi-omics and cross-modal cellular dynamics.
Conclusion
The scCross method offers significant potential for the single-cell research community, addressing challenges that may be difficult to overcome with existing approaches. Its unique features and reliable performance make it a valuable tool for researchers engaged in single-cell multi-omics analysis. scCross facilitates the integration of different modalities, supports comprehensive data generation, and enables detailed simulation and perturbation, which could advance the study of complex biological systems. We encourage researchers to explore scCross and consider its application in their studies. For further details, please refer to our paper in Genome Biology (https://doi.org/10.1186/s13059-024-03338-z).
Follow the Topic
-
Genome Biology
This journal publishes outstanding research in all areas of biology and biomedicine studied from a genomic and post-genomic perspective.

Related Collections
With Collections, you can get published faster and increase your visibility.
Tackling large-scale genomic studies
Genome Biology is calling for submissions to our Collection on large-scale genomic studies. Modern genomic research now generates datasets of unprecedented scale spanning population cohorts, large single‑cell atlases, and high‑throughput multi‑omics studies. These expansive datasets offer powerful opportunities to uncover biological mechanisms, but they also introduce major challenges in data management and interpretation. Addressing these demands requires scalable approaches capable of extracting meaningful insight from large and heterogeneous genomic resources.
This Collection invites contributions that advance the design, execution, and interpretation of large‑scale genomic studies, including:
- Experimental and sequencing strategies optimised for high‑throughput, population‑scale data generation
- Frameworks for data harmonisation and standardisation, enabling cross‑study comparability, meta‑analysis, and integration of datasets generated across platforms, cohorts, or populations
- Scalable machine learning and AI approaches designed for high‑throughput genomic data
- New research with biological insights derived from large‑scale genomic analyses
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Dec 05, 2026
Prioritizing non-coding variants
Genome Biology is calling for submissions to our Collection highlighting methodological advances and the application of those methods that enable identification, annotation, and biologically meaningful interpretation of non-coding variants.
Non‑coding variants are changes in genomic regions that do not encode proteins but may influence gene regulation, RNA biology, chromatin structure, or other functional genomic processes. Despite their importance, the functional interpretation of non‑coding variants remains one of the most significant challenges in genomics. The complexity, context‑dependence, and subtle phenotypic effects of these variants require innovative experimental and computational strategies to resolve regulatory mechanisms across diverse biological systems.
This Collection invites contributions at the intersection of genomics, regulatory biology, and computational modelling that advance the prioritization and functional interpretation of non‑coding variants, including:
- Experimental approaches for identifying variants in non-coding regions, perturbing non-coding sequences, or characterising variant effects
- Computational frameworks for predicting the regulatory function of non-coding variants in gene regulation, RNA process, chromatin structure, or other biological processes
- Methods for integrating diverse data types, including epigenomic, transcriptomic, spatial, and 3D genome datasets, to improve variant prioritisation and mechanistic interpretation
- Benchmarking studies that evaluate the performance and reproducibility of tools for non‑coding variant identification or annotation
- Translational applications linking non‑coding variants to disease mechanisms, therapeutic targets, pharmacogenomic responses, or genotype–phenotype relationships
Submissions that represent a substantial advance over previous studies—whether through methodological innovation, comprehensive benchmarking, or generation of new biological insight into the regulatory mechanisms of non‑coding variants—are especially encouraged.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Nov 25, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in