Modeling metal biointerfaces and electrodes up to millions of atoms
Published in Chemistry
The paper in Nature Communications is here: http://go.nature.com/2sFKoJz
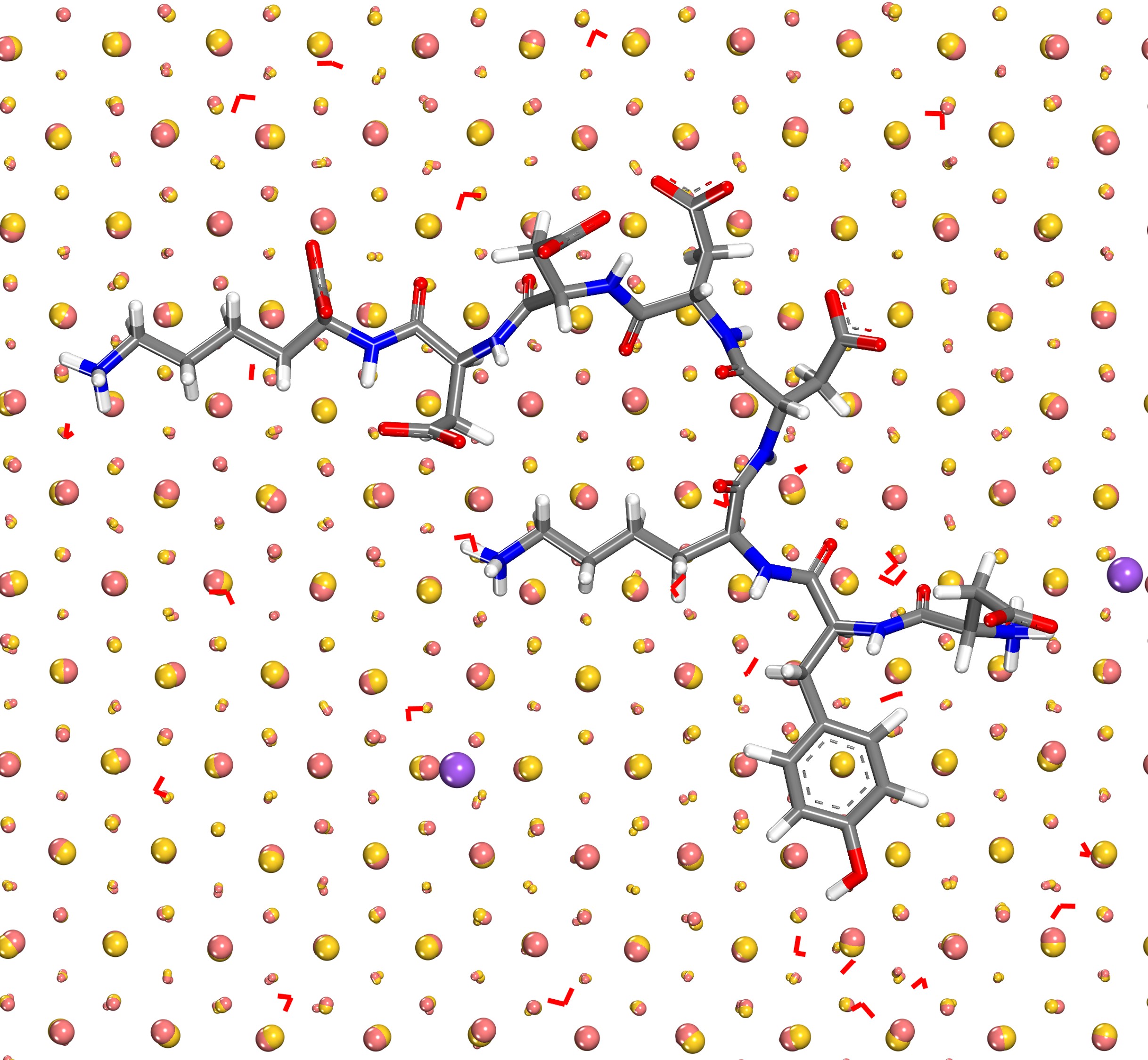
Nanostructured metals and alloys are great materials for next generation catalysts, sensors, and therapeutics. The synthesis usually involves complex solution chemistry and poses many open questions. Density functional theory (DFT) is often engaged in computations as it is easy to apply for any composition, however, DFT comes with limitations in scale to a few hundred atoms and with results of mixed reliability that is often not well documented. Computed surface and interfacial energies of metals deviate from experimental measurements between 30% and 70% using common density functionals. The polarizable Lennard-Jones potential introduced here reduces these deviations to less than 5%, electrical polarization in response to external charges and fields is included, and a million more systems can be studied per unit time. Computed binding energies of water monolayers agree with experimental measurements with under 1 kcal/mol deviation and binding of biomolecules can be studied in chemical accuracy. The simulation of complex systems up to 100 nm size is feasible without adding any further parameters for interfacial interactions.
The adsorption of electrolytes, biopolymers, gases, and oxides on the metal surface can thus be modeled in high precision and extended to simulate surface reactions more accurately than current reactive force fields and DFT methods allow. Reaction simulations with DFT are difficult to rely on as uncertainties around 50% in surface properties eliminate essential differences between metals, for instance, it is not possible to distinguish surface properties of aluminum, silver, and gold which have surface energies of 1.18, 1.32, and 1.54 J/m2, respectively. The new force field, in combination with experimental data and quantum mechanical calculations has a better chance to predict small differences in activation energies on the order of 1 to 3 kcal/mol that can make the difference between a commercially viable and a non-starter catalyst.
The study further illustrates that the attraction of ions to a metal surface in vacuum as a result of induced charges can be as strong as a chemical bond while at a solution-metal interface or an oxide-metal interface the impact of induced charges is far smaller. Our results disprove earlier assumptions of a strong, or dominant, influence of induced charges in condensed matter-metal interfaces. Induced charges do affect ionic solutions or minerals in contact with metals, however, the magnitude is only equal to intermolecular bonds and can now be included on the fly in molecular simulations. The assembly of soft matter such as biomacromolecules and polyelectrolytes on metal surfaces is therefore affected by induced charges although the magnitude is much less than ionic interactions.
We also found it helpful to associate parameters in models with a physical interpretation, beginning with understanding the chemistry of the system and translating the key features into models. Computer models that represent a basic “molecular code” allow direct connections between these essential atomic (or electronic) scale characteristics and simulation results, making rational design more facile. In this model, Lennard-Jones parameters encode the density and surface energy while charges of a stretchable dipole represent polarizability and (via their magnitude) mechanical properties. In contrast, models with merely arithmetic fit parameters and a large number of parameters (DFT, some reactive FFs) are often used as a black box. Results are difficult to trace back to underlying assumptions and interpretations no matter whether the results are of high or low quality. For research purposes, I recommend models we can understand and improve. If they are also good for making predictions, they might prevail.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in