MultiPro: a comprehensive proteomic resource for investigation of multiple technical issues
Published in Protocols & Methods and Research Data
Multitudinous technical issues in proteomics data
Mass spectrometry (MS)-based proteomics has important applications for biomedical research in the areas of phenotype correlation and characterization1, biomarker and drug discovery2,3, and therapeutics4. Although powerful, proteomic data are challenging to generate, use and study. In practice, proteomics is not a single technology but comes in many flavours and platforms. For example, proteomics can be pursued via different data acquisition modes, e.g., data-dependent acquisition (DDA) and the more recent data-independent acquisition (DIA) coupled with various liquid chromatography (LC) systems and quantitation strategies (e.g., labelled and label-free). This diversity creates challenges for cross-platform data integration5. The non-amplifiable nature of proteins and instrument resolution limits lead towards missing values, especially amongst low-abundant proteins6. Moreover, instrument throughput issues mean samples often need to be batched across various machines and laboratories. This can produce challenging technical biases known as “batch effects”7–9. Recent works have demonstrated co-dependence and interactions between these issues, i.e., between missing value imputation (MVI) and batch effect correction (BEC)10. These issues can confound true biological signals, reduce reliability of downstream statistical analysis, and lead towards research outcome irreproducibility5,6,8.
Golden benchmark datasets are crucial but scarce
To deal with the above issues, it is insufficient to simply apply general algorithms or borrow established methods from similar domains. It is better to develop specific algorithms for the platform. But to achieve this, we need appropriate benchmark datasets for algorithm development and evaluation.
However, current datasets available on public repositories may be unsuitable for deep diving into such investigations. Firstly, most publicly available datasets were originally designed or intended for answering specific research question rather than for benchmarking, which means these datasets comprise mostly biological replicates from different individuals like patients or mice, resulting in higher heterogeneity than needed7,11.
Another key limitation is the lack of cross-platform datasets. Most clinical proteomic datasets were only acquired through one platform, and cannot be used for investigating, comparing, and integrating across proteomics platforms for the purpose of understanding platform interoperability, data transformation and integration, or modelling platform-specific technical biases.
Finally, there is a lack of benchmark datasets derived from new promising technologies such as PASEF (Parallel Accumulation-Serial Fragmentation), which has been developed to provide significant gains in sensitivity, coverage and reproducibility12.
MultiPro: a comprehensive proteomic resource for investigation of multiple technical issues
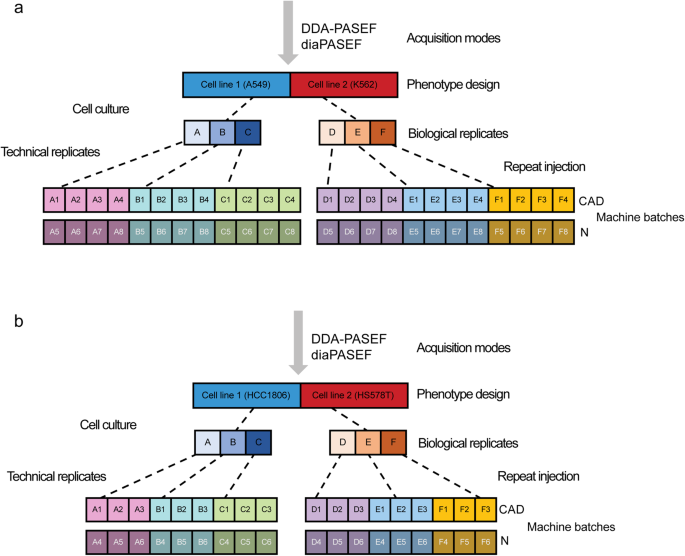
We present MultiPro (Multi-purpose Proteome Resource)13, which comprises a suite of four comprehensive large-scale proteomic datasets with deliberate design. The samples were generated in DDA-PASEF14 or diaPASEF15 mode using standard data processing workflows16. MultiPro contains a balanced two-class design based on well-characterized cell lines (A549 vs K562 or HCC1806 vs HS578T), with controlled batch effects established using two different MS instruments and 48 or 36 biological and technical replicates altogether in each dataset (Figure 1).
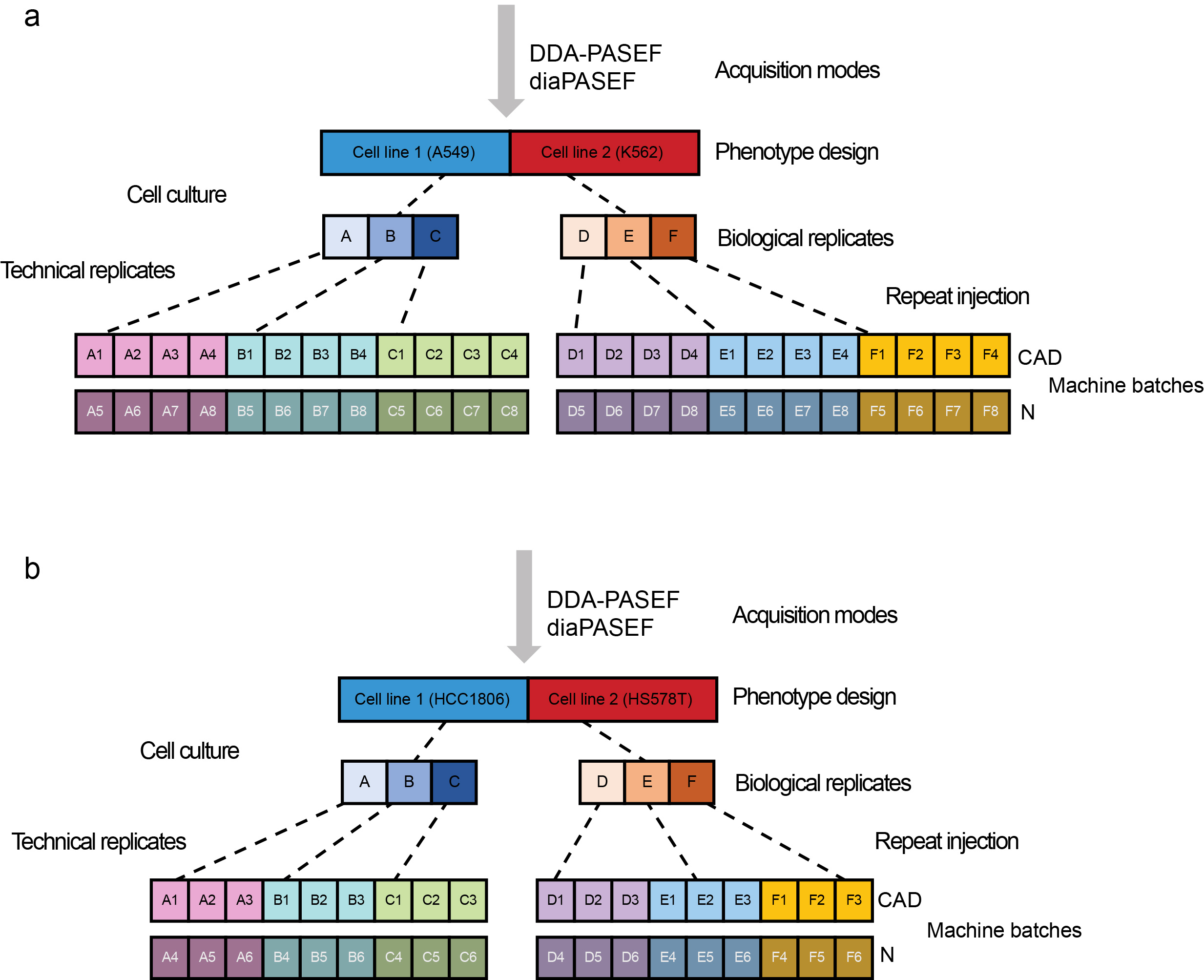
Figure 1: Experimental design of MultiPro. (a) A549/K562 experimental design. (b) HCC1806/HS578T experimental design. This figure was adapted from the original paper13 (https://www.nature.com/articles/s41597-023-02779-8).
Benefits of using MultiPro
Wide-ranging and precise proteome identification and quantification
With deliberate experimental design and standard data generation pipeline, MultiPro not only offers a holistic and robust view of identification and quantification per sample but also highlights significant variations across samples, while ensuring steadfastness across technical replicates.
Missing pattern differs by acquisition method, class and batch
Various missing structures and mechanisms from different MS methods, classes and batches have been constructed in MultiPro, making it a suitable resource for investigating missing value issues and providing useful scenarios to evaluate the performance of MVI methods especially when missing values interact with batch or class effects (Figure 2).
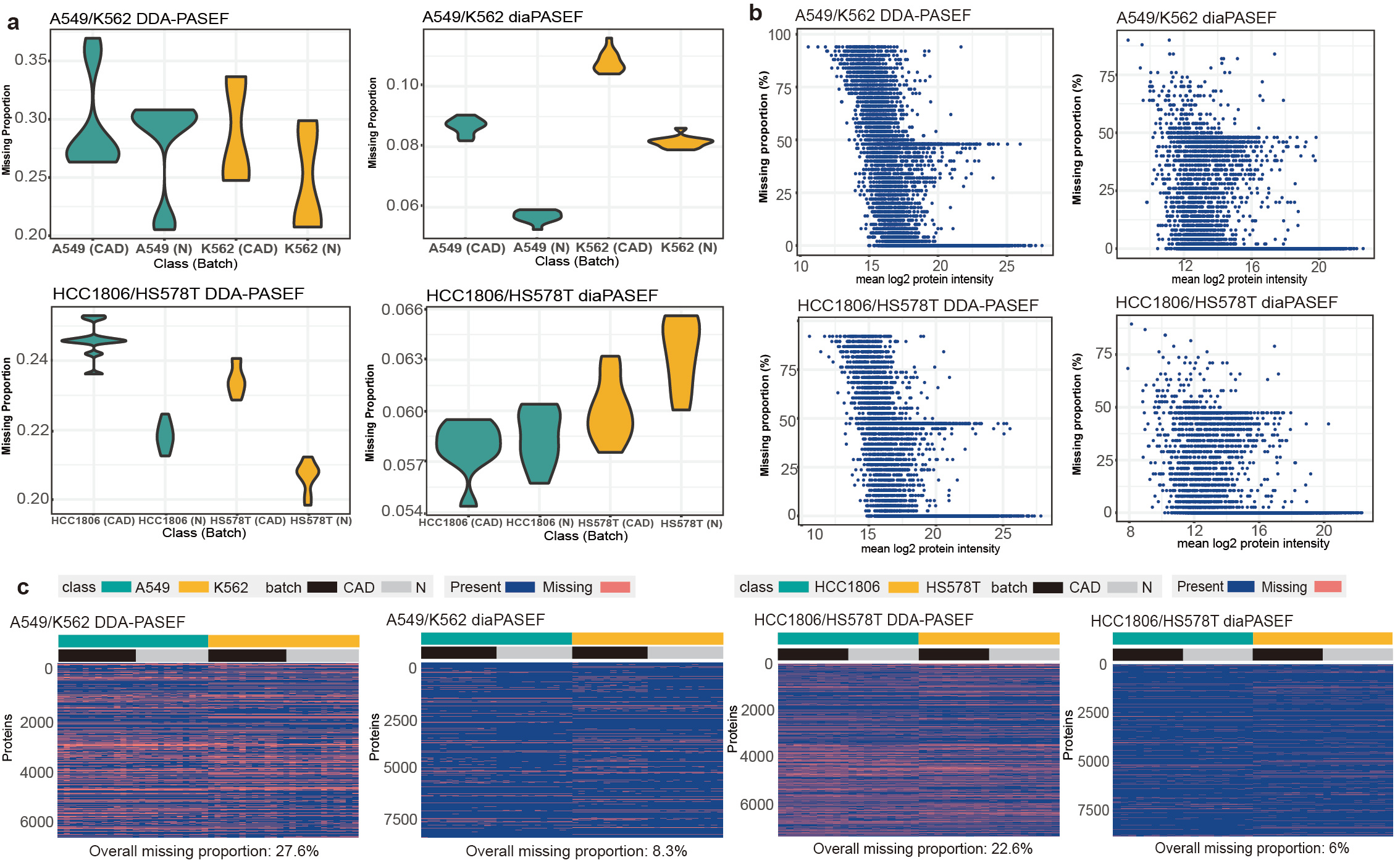
Figure 2: Various missing patterns of MultiPro. (a) Violin-plots representing missing proportions of samples categorised by class and batch. (b) Scatterplots displaying missing rates of proteins ordered by mean log2 intensity. (c) Heatmaps demonstrating missing structures and patterns. This figure was adapted from the original paper13 (https://www.nature.com/articles/s41597-023-02779-8).
Mixed effects from class, batch and acquisition mode
Furthermore, using visual and quantitative bioinformatics tools, mixed effects from class, batch and acquisition method can be demonstrated and investigated through MultiPro, which helps the development and evaluation of more powerful batch effect correction algorithms (Figure 3).
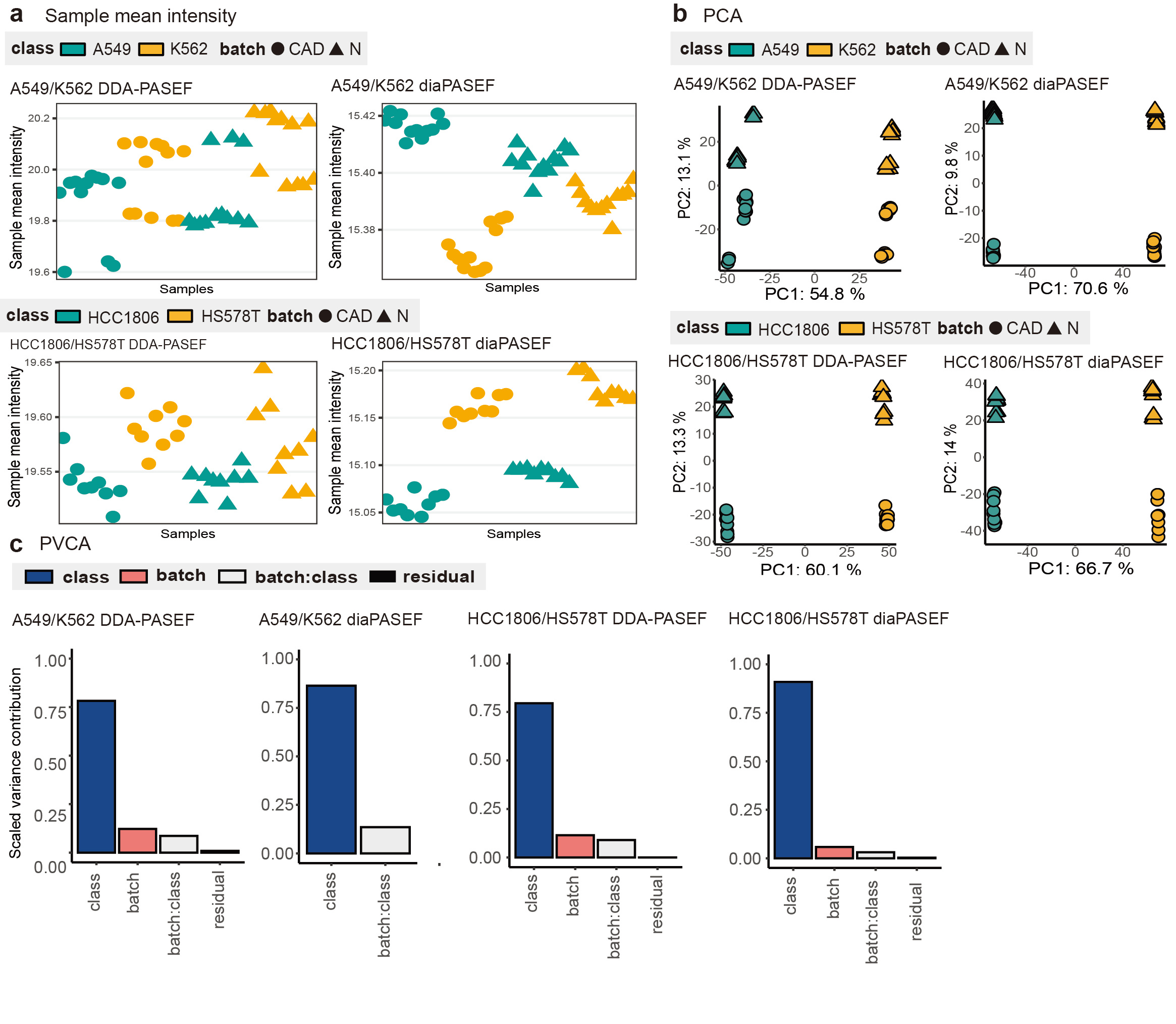
Figure 3: Batch effects demonstration of MultiPro. (a) Scatterplots demonstrating the mean intensities of samples. (b) Principal Component Analysis visualization. (c) Principal Variance Component Analysis. This figure was adapted from the original paper13 (https://www.nature.com/articles/s41597-023-02779-8).
Use cases for MultiPro
- The comprehensive design of MultiPro not only allows for investigation of a multitude of technical issues, including cross-platform data integration, missing value imputation, batch effects correction and their inter-connections, but also enables development and benchmarking of bioinformatic algorithms.
- Direct DDA-PASEF and diaPASEF pairings per sample allow for meaningful validation of predicted missing proteins/values. These pairings are also useful as validations when attempting to use generative artificial intelligence model to transform data from one platform type to another, allowing for advanced applications to improve cross-platform inter-operability and facilitating data integration.
- Given the present batch factors, investigators can also evaluate BEC algorithms in terms of the consistency of returned differential proteins, or the consistency of results across batch/class pairs.
- MultiPro can interact with other published datasets17,18 which also quantified the same cell lines as ours (A549, K562, HCC1806 and HS578T) but using diverse data generation paradigms across various time periods, to mimic different kinds of batch effects (e.g., same instrument but different days, instruments across different labs, different LC columns) and benchmark BEC algorithms thoroughly.
References
- Jayavelu, A. K. et al. The proteogenomic subtypes of acute myeloid leukemia. Cancer Cell 40, 301-317.e12 (2022).
- Niu, L. et al. Noninvasive proteomic biomarkers for alcohol-related liver disease. Nat. Med. 28, 1277–1287 (2022).
- Meissner, F., Geddes-McAlister, J., Mann, M. & Bantscheff, M. The emerging role of mass spectrometry-based proteomics in drug discovery. Nat. Rev. Drug Discov. 21, 637–654 (2022).
- Jiang, Y. et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 567, 257–261 (2019).
- Santos, A. et al. A knowledge graph to interpret clinical proteomics data. Nat. Biotechnol. 40, 692–702 (2022).
- Kong, W., Hui, H. W. H., Peng, H. & Goh, W. W. B. Dealing with missing values in proteomics data. PROTEOMICS 22, 2200092 (2022).
- Čuklina, J. et al. Diagnostics and correction of batch effects in large-scale proteomic studies: a tutorial. Mol. Syst. Biol. 17, e10240 (2021).
- Goh, W. W. B., Wang, W. & Wong, L. Why Batch Effects Matter in Omics Data, and How to Avoid Them. Trends Biotechnol. 35, 498–507 (2017).
- Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 11, 733–739 (2010).
- Hui, H. W. H., Kong, W., Peng, H. & Goh, W. W. B. The importance of batch sensitization in missing value imputation. Sci. Rep. 13, 3003 (2023).
- Sundararaman, N. et al. BIRCH: An Automated Workflow for Evaluation, Correction, and Visualization of Batch Effect in Bottom-Up Mass Spectrometry-Based Proteomics Data. J. Proteome Res. 22, 471–481 (2023).
- Wang, H. et al. MultiPro: DDA-PASEF and diaPASEF acquired cell line proteomic datasets with deliberate batch effects. Sci. Data 10, 858 (2023).
- Meier, F. et al. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteomics MCP 17, 2534–2545 (2018).
- Meier, F. et al. Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 14, 5378–5387 (2015).
- Meier, F. et al. diaPASEF: parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods 17, 1229–1236 (2020).
- Cai, X. et al. High-throughput proteomic sample preparation using pressure cycling technology. Nat. Protoc. 17, 2307–2325 (2022).
- Gonçalves, E. et al. Pan-cancer proteomic map of 949 human cell lines. Cancer Cell 40, 835-849.e8 (2022).
- Sun, R. et al. Proteomic Dynamics of Breast Cancer Cell Lines Identifies Potential Therapeutic Protein Targets. Mol. Cell. Proteomics 22, (2023).
Follow the Topic
-
Scientific Data
A peer-reviewed, open-access journal for descriptions of datasets, and research that advances the sharing and reuse of scientific data.

Related Collections
With Collections, you can get published faster and increase your visibility.
Computer vision in plant science and agriculture
Publishing Model: Open Access
Deadline: Oct 10, 2026
Wearable and Computer Vision Data for Health and Behaviour Research
Publishing Model: Open Access
Deadline: Aug 08, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in