Neutrophils play an unexpected role in bioengineered vasculature
Published in Bioengineering & Biotechnology

The discovery of non-inflammatory neutrophil involvement in revascularization is quite literally a long story. When I first joined Juan Melero-Martin’s lab as a postdoc in 2009, I was immediately interested in building human blood vessels for regenerative medicine. We found that two essential cellular components are required for the creation of blood vessels. One is the endothelial cells that line the inner lumen of blood vessels. The other is the perivascular cells surrounding endothelial tubes for vessel stability. Both human cells could be derived from feasible sources of postnatal progenitors. The endothelial colony-forming cells (ECFCs) are endothelial progenitors with robust proliferation and vessel-forming potential that can be isolated from both adult and umbilical cord blood. On the other hand, mesenchymal stem cells (MSCs), which are abundant in bone marrow and adipose tissue, provide a reliable source for perivascular partners. When we mixed these two cell types together in a hydrogel and injected them into the subcutaneous space on the back of hairless immunodeficient mice, these cells self-arrange and assemble into functional human vessels through vasculogenesis, a process where a primitive vascular bed forms during embryonic development. These human micro-vessels become wired into an extensive vascular network and connected to host mouse circulation, meaning you can see mouse red blood cells flowing inside the human lumen. The entire process from injected single cells to perfused vasculature takes only 5-7 days. The ability to functionally engraft a human vasculature in an in vivo animal model opens various applications from cell therapy for ischemic tissue to synthetic protein delivery.
We then wondered what was happening in the small space during blood vessel formation. We performed daily observations and found that a wave of myeloid cells infiltrate the implants soon after cell injection and gradually fade away simultaneously with blood vessel formation. When this myeloid population is depleted by specific antibodies, the formation of human blood vessels is largely impaired, suggesting that the leukocyte infiltration is not necessarily a bad thing. Some host cells may play an important role in facilitating the vascularization of human vascular cells.
I tried to narrow down the key players to two major subtypes of myeloid cells, macrophages and neutrophils. Neutrophils enter the vessel-forming area within 2 hours post injection followed by macrophages with a 24-hour delay. The number of infiltrated cells peak at day 2 and then neutrophils gradually disappear, but macrophages maintain a consistent population even after vessel formation. Like any rookie new to this field, I started digging into the literature on Pubmed. At that time, M2 macrophages were under the spotlight for their unique role in tissue remodeling. Inevitably we formulated a hypothesis that macrophages are recruited to build the blood vessels, meanwhile neutrophils just come to clean the dead cells.
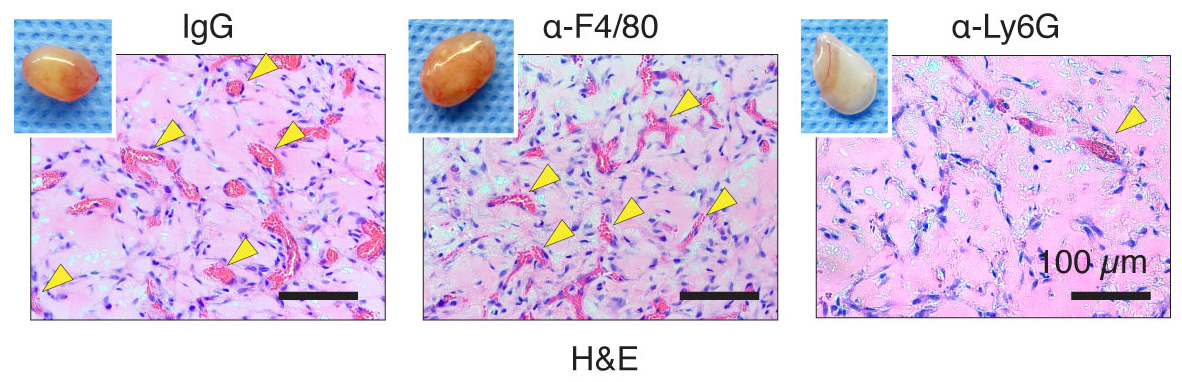
With this concept in mind, I did a selective monocyte depletion using an antibody against a monocyte/macrophage marker, F4/80. The depletion worked beautifully to wipe out the entire monocyte population from mouse blood, meaning the major source of recruited macrophages is empty. “As a control”, neutrophils were depleted as well by anti-Ly6G antibody. When another postdoc in the lab, Rafael, and I opened the mouse skin and saw the well-vascularized plug in F4/80-treated mice, we immediately double-checked the mouse cage cards to make sure we didn’t operate on the wrong groups. Soon after, we opened the Ly6G-treated mice and found a pale, lifeless gel, and again couldn’t believe the results. Two weeks later, we repeated the experiments and saw the exact same results. Due to the failure of our hypothesis we began to consider abandoning the project and moving in a different direction.
Curiosity brought us to revisit this puzzle a few months later. This time, we changed our approach by depleting all white blood cells from mouse blood by sublethal doses of irradiation, and then performing adoptive cell transfer from a healthy donor to reconstitute specific subtypes. Again, adoptive transfer of neutrophils, but not monocytes, rescued the vessel formation. Now, we knew we needed to take this seriously but instead focusing on the importance of neutrophils.
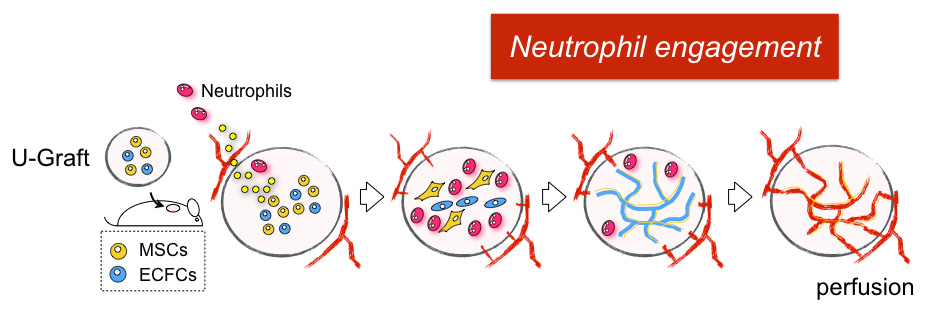
This is the story behind our paper. We hope it adds to your enjoyment of the article. In summary, we found human vascular cells actively recruit neutrophils through the secretion of neutrophil chemokines, including IL6, CXCL1 and CXCL8. Once neutrophils enter the vessel-forming area, TGF-b, which is also secreted from human cells, directs them toward alternative activation of “N2” neutrophils. These N2 neutrophils display a distinct non-inflammatory phenotype with upregulation of anti-inflammatory and tissue remodeling genes. Both depletion of neutrophils and blocking of TGF-b signaling results in impaired vascularization. The ability of human vascular cells to recruit neutrophils was progressively silenced as the vasculature matured. This explains why the implantation of assembled human vascular networks (for example, a tissue-engineered construct or a piece of real tissue) fails to engraft, but the same cells in a hydrogel suspension form vascularization. We also found the progressive loss of neutrophil engagement is regulated by the Notch pathway, which is known to govern vascular maturation. Blocking Notch signaling reactivates the neutrophil program and significantly improves the vascularization of our mature vascular bed. Looking forward, we expect this new role of non-inflammatory neutrophils will attract broad attention for their potential benefit on improving surgical grafting and tissue regeneration.
Although our initial hypothesis was proven completely wrong, our scientific curiosity led us from defeat down the path to discovery and that is the nature of research.
Our paper: Lin, R.-Z. et al. Host non-inflammatory neutrophils mediate the engraftment of bioengineered vascular networks. Nat. Biomed. Eng. 1, 0081 (2017).


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in