New ways to control blood vessel growth in eye disease
Published in Chemistry and Anatomy & Physiology
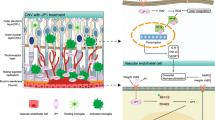
When we began this project, the hypothesis felt attractively simple. Endothelial cells line blood vessels, and in disease they are often asked to do contradictory things: remain stable enough to preserve vascular integrity, but flexible enough to sprout, remodel and respond to injury.
ZEB1 is a zinc-finger E-box binding transcription factor best known for regulating cell plasticity. In epithelial tissues, it is strongly associated with epithelial-to-mesenchymal transition, where it represses epithelial junctional genes, promotes migratory behaviour and helps cells acquire more flexible, stress-adapted states. It has also been linked to stemness, fibrosis, immune regulation and cancer progression. Although much of this biology has been studied in epithelial or tumour cells, many of the same themes are highly relevant to endothelial cells. Endothelial cells also need to alter their identity during development, inflammation and pathological angiogenesis (the growth of blood vessels), while preserving vascular integrity. This made ZEB1 an attractive candidate for controlling the balance between endothelial activation, plasticity and barrier function.
Our starting idea was that endothelial ZEB1 might help drive pathological vascular remodelling. The first clues came from publicly available transcriptomic datasets showing that Zeb1 was not randomly expressed across endothelial populations. It was enriched in activated and remodelling endothelial cells, including those associated with choroidal neovascularisation. The question was whether it was simply a marker of endothelial activation, or whether it was doing something more instructive. In some regards we still do not fully know, but the data suggested that it was worth testing functionally.
Choroidal neovascularisation, or CNV, became an especially useful setting in which to ask that question. CNV is a powerful model of pathological angiogenesis because it captures several linked features of disease-associated vascular growth: endothelial activation, neovascular invasion, vascular remodelling and leakage. It is not a perfect replica of human age-related macular degeneration, but it provides a robust experimental system in which new vessels form in a tissue context where vascular dysfunction has clear biological and clinical meaning. Importantly, it allowed us to think about pathological angiogenesis not only as a question of whether vessels grow, but whether they become stable, leaky, inflamed or functionally abnormal. That distinction became central to the paper.
We initially expected endothelial ZEB1 loss to blunt angiogenic behaviour. Instead, in vivo deletion increased aspects of neovascular growth and leakage. In the laser-induced CNV model, loss of endothelial ZEB1 enhanced vascular invasion and vascular leakage, yet did not produce the inflammatory phenotype we might have anticipated. That was the first moment where the project forced us to slow down. Increased pathological vascular growth, increased leakage, but not a straightforward increase in inflammation. It was a more selective endothelial phenotype than our original model allowed.
This is where the computational work became particularly useful. Public single-cell datasets gave us a way to ask not only where Zeb1 was expressed, but how its expression might relate to broader endothelial cell states and regulatory programmes. Using a a computational biology approach, we modelled how gene regulatory networks might be altered when ZEB1 activity is disrupted. Rather than looking at single genes in isolation, this allowed us to ask a more systems-level question: if ZEB1 changes, which parts of the endothelial programme are predicted to shift with it?
The predictions were intriguing. They suggested that ZEB1 could be linked to inflammatory and activation-associated signalling within the most angiogenic endothelial populations. More accurately, the modelling suggested that disrupting ZEB1 might be expected to disturb the inflammatory behaviour of these endothelial cells. This was helpful because our experimental phenotype was not a simple “more inflammation” or “less inflammation” story. The knockout vessels were more invasive and more leaky, but they did not show the broad inflammatory increase we had expected. The computational analysis helped us think about this as a rewiring of endothelial state rather than a uniform gain or loss of inflammation. It helped generate a more refined interpretation of the experimental data. It suggested that ZEB1 might sit within a regulatory network that helps endothelial cells coordinate angiogenesis, barrier function and inflammatory responsiveness. If that network is disrupted, the resulting vessels may grow and leak, but fail to activate inflammatory pathways in the way one might predict from vessel damage alone.
The ability to combine this model with non-invasive imaging was also particularly important. Rather than relying only on endpoint measurements of lesion size or vessel area, we could ask how the vasculature behaved functionally in vivo. Leakage matters because it changes the meaning of angiogenesis. A new vessel that is stable and integrated is biologically different from one that is fragile, permeable and disruptive to the surrounding tissue. Non-invasive imaging helped us place vascular function alongside vascular structure, and that made the phenotype harder, but also more interesting, to interpret.
The in vitro work added another layer. Knocking down ZEB1 in endothelial cells disrupted leukocyte adherence, which we also saw in tissue isolated from the knockout mice. Other work from our laboratory has shown that ZEB1 can also influence the expression of proteins that contribute to endothelial barrier formation and permeability. These experiments were technically simpler than the animal work, but conceptually they were crucial. They suggested that ZEB1 was not merely suppressing or promoting angiogenesis in a broad sense. It appeared to help endothelial cells maintain a competent barrier state while navigating activation. Removing ZEB1 did not just make vessels “less angiogenic” or “more angiogenic”; it altered the quality of the endothelial response.
Like many projects that were live during the COVID period, this work was heavily affected by disruption. Experiments that should have followed each other cleanly became fragmented. Access to facilities, animal work, cell culture, imaging and analysis all had to be reassembled around changing restrictions, staff availability and the practical realities of keeping research moving. Momentum was not something we could take for granted. There were long periods where the main job was not generating new data, but protecting the logic of the project: remembering why each experiment mattered, what the previous result had actually shown, and what interpretation was still justified.
In some ways, that enforced slowness helped the science. It made us less willing to flatten the data into the story we first wanted to tell. The in vivo and in vitro data did not simply confirm each other in a tidy, linear way. They had to be held together carefully. The animal model showed that endothelial ZEB1 loss altered pathological neovascularisation and leakage. The cell work pointed toward a role in endothelial stability, leukocyte interaction and barrier regulation. The public transcriptomic data placed Zeb1 within activated endothelial states. The computational modelling suggested that ZEB1 disruption could reshape inflammatory and angiogenic endothelial programmes. None of these alone would have been enough. Together, they pushed us toward a more nuanced view: endothelial ZEB1 acts less like a simple on/off switch for angiogenesis, and more like a regulator of endothelial phenotype during vascular stress.
This also made us reflect on other endothelial ZEB1 stories emerging from the lab. Across different vascular beds and disease models, ZEB1 kept appearing at points where endothelial cells were under pressure to change state. In some contexts, ZEB1 seemed connected to vascular remodelling. In others, it appeared more closely linked to barrier function, leukocyte interaction or developmental progression. These were not identical phenotypes, and that is precisely what made them useful. They suggested that ZEB1 is not imposing one universal endothelial programme. Instead, its role depends on context: tissue, vascular bed, developmental stage, injury state and the demands being placed on the endothelium.
The most interesting biology was not that ZEB1 “causes” or “prevents” angiogenesis. It was that ZEB1 helps shape the type of endothelial response that emerges during pathology. In the choroid, where vascular leakage is central to disease relevance, this becomes particularly important. A vessel can grow, but the consequences of that growth depend on whether it is stable, leaky, inflamed, perfused or functionally integrated. Our data suggest that endothelial ZEB1 contributes to this qualitative control.
There were also broader lessons about how we use models. The laser-induced CNV model gave us a reproducible vascular injury and neovascular response, but it still needed to be interpreted through complementary approaches. Publicly available datasets allowed us to ask where Zeb1 sits within endothelial heterogeneity, rather than treating the endothelium as a single uniform compartment. Gene regulatory netwoek modelling allowed us to ask how ZEB1 might be connected to wider regulatory programmes. In vitro assays let us isolate endothelial-intrinsic mechanisms, but could not recreate the full tissue environment. Non-invasive imaging allowed us to connect structural angiogenesis to functional vascular behaviour. The value came from allowing each system to challenge the others.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in