On the hunt for the non-enzymatic Krebs cycle
Published in Ecology & Evolution
Behind the paper: On the hunt for the non-enzymatic Krebs cycle
Metabolism is a universal feature of life. As biochemists we are used to, and barely question, the fact that all biological systems depend on a highly similar (although not identical), set of key metabolites and metabolic pathway reaction sequences. Indeed, we name metabolic pathways differently if they differ in just one or two steps (for instance, I did once trigger a highly emotional response from a reviewer at Science, by referring to both the (substantially overlapping) Entner-Doudoroff and the Embden-Meyerhof pathways as ‘glycolysis’). In fact, you can easily confuse about half of the audience in a Biochemistry seminar if you use the term ‘conserved’ to describe anything other than a nucleotide or amino acid sequence, referring to a pathway’s reaction topology instead: we are very comfortable with the idea that the amino acid sequence of enzymes differs between species, but take it almost for granted that the network structure which connects these enzymes is more or less unchanged all the time.
Yet, reflecting from a chemist’s point of view, the conservation of metabolic reaction sequences is anything but trivial. Chemistry offers hundreds of different possibilities, for example, to form pyruvate out of glucose, as cells do through glycolysis. Why phosphorylating it, and why is the phosphorylation occurring almost universally on the 6th carbon position before glucose can be converted into anything else? This is a requirement of biology, not chemistry. The typical textbook replies to this problem with Darwinian logic - surely glucose phosphorylation on C6 must be the most ‘optimal’ solution? But life on Earth persist (even thrives) in a tremendous diversity of environments: some thermophilic Archaea are quite happy living at 80°C in oxygenated hot-springs at a pH of 2 (Brock et al. 1972), while at the same time snow fleas spend their days using a substantial amount of their limited carbon and energy producing anti-freeze proteins in order to avoid the growth of ice crystals (Graham and Davies 2005). All these environments require the production of metabolites at different quantities. For all the different conditions and different metabolic requirements, the exact same chemistry always the most optimal solution? I doubt it.
More likely, evolution doesn’t allow change of metabolic reaction sequences very easily. At first, the evolutionary origin of a metabolic pathway inevitably creates a chicken-egg problem if there is no chemical template to build upon (without product there is no evolutionary advantage in Darwinian terms, but to form the product you may need pathway intermediates first. However, these provide no advantage unless the product in subsequently formed (Horowitz 1945)). So new pathways do not come into place out of nothing, and for sure not step-wise. Second, novel reactions need to be compatible with the existing metabolic system. Ask metabolic engineers how difficult this is! The supposedly trivial task to add an efficient metabolic pathway to form artemisinic acid in yeast took about ~150 postdoc-years to work efficiently (Ro et al. 2006).
Hence, these considerations put into question whether the core parts of the metabolic network structure have changed much over the evolutionary timeline. A few years ago we stumbled across a potential solution to this problem. In a control experiment a talented postdoc in my lab, Markus Keller, heated-up metabolites of glycolysis to test whether they could account for some pyruvate that we discovered as contaminant in our yeast media after autoclaving, a process of heat sterilisation often used by microbiology labs. The heat exposure did indeed account for the pyruvate formation. Furthermore, we realized that glycolytic intermediates do not just form pyruvate, but interconvert also other intermediates of glycolysis. Is it possible that the structure of the metabolic network is determined by non-enzymatic reaction properties of its intermediates, instead the consequence of a Darwinian optimisation process that selected for metabolic pathways by providing the enzymatic catalysts? This hypothesis being correct, it would propose that the chemical environment of early organisms must have had a crucial in impact on the shape of the prevailing metabolic network structure.
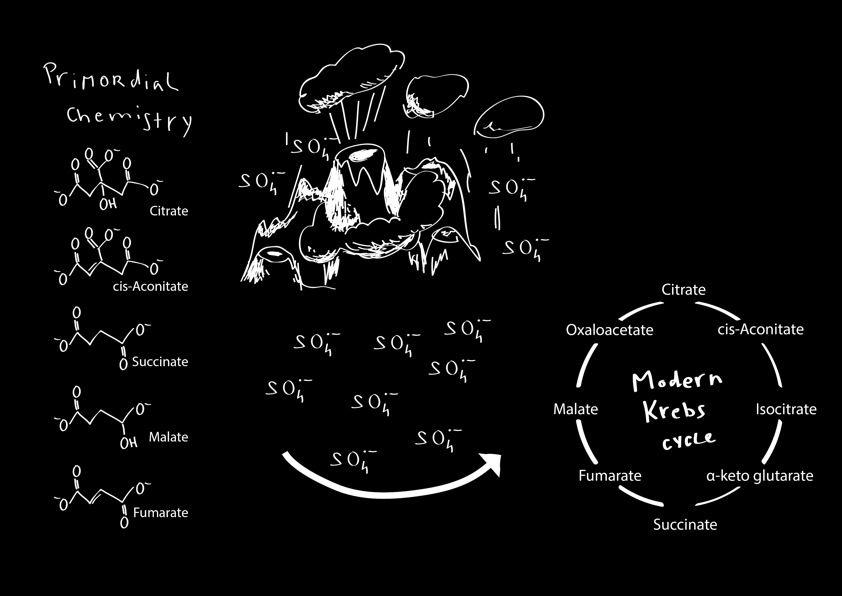
Not knowing much about the time at which the origin of metabolism most likely occurred, I sent a request to the Earth Science Department at Cambridge University. Explaining my problem, I was brought into contact with Sasha Turchyn, an expert of Archean oceanic sediments. Together, the three of us designed a large systematic test series, adding the typical components of Archean sediment to observe their impact on intermediates of glycolysis. Markus got one hit, and much to the Geoscientist’s excitement, this was the most highly concentrated metallic sediment component, ferrous iron. Fe(II) is present in huge quantities in aquatic environments on the pre-oxygenated world owing to its solubility in water. It enabled a large spectrum of additional reactions among glycolytic intermediates, as well as increasing the efficiency and speed of the chemical reaction network among the metabolic intermediates. In other words, Fe(II) driven interconversion reactions among sugar phosphates form two chemical networks that Biologists know under two names: glycolysis and the pentose phosphate pathway (PPP) (Keller et al. 2016; Keller, Turchyn, and Ralser 2014; Luisi 2014).
While most origin of life researchers do not think often about the origin of the topological structure of the extant metabolic network (as said, most of us consider the chemical structure very much a constant), they think a lot more about another important question - whether early life was of heterotrophic or autotrophic nature. We didn’t address this problem in our work, but admittedly have included some reference to this problem in our 2014 manuscript when reporting for the first time about the problem (in essence, as one Reviewer asked us to do so). The consequence was that some of our readers confused non-enzymatic reactions that replicate the glycolytic topology with a claim that life was of heterotrophic origin, and that we would fancy an Archean ocean full of sugar phosphates. Although we did not make such claims (least not even addressed the heterotrophy/autotrophy problem!), we were heavily criticized by some of our colleagues for our supposedly defence of such primoridal soup theory, in both web-blogs or scientific papers ((Schönheit, Buckel, and Martin 2016), http://sandwalk.blogspot.co.uk/2014/05/more-primor...). However, the particular critique made against the importance of non-enzymatic reactions is relatively easy to rebut, as the laws of thermodynamics do not disappear when considering enzymatic reactions instead of their non-enzymatic counterparts. The fact that glycolysis and the PPP operate in almost all cells is a hard-to-deny proof that evolution did overcome these thermodynamic barriers, despite the fact no ocean was ever full of sugar phosphates. In other words, if a non-enzymatic glycolysis would be a thermodynamic ‘nonsense’, an enzymatic glycolysis would equally nonsensical, with the consequence that the writers from both sides of the debate would die in an instance from a lack of ATP.
One fact is, however, certainly true. Chemical (this includes also enzymatic!) networks develop towards the thermodynamic equilibrium. In order for a metabolic system to be able to emerge, both anabolic ('breaking-up' ) reactions and catabolic ('building-up') reactions need to co-occur, otherwise the system would cease once the equilibrium is reached (in essence, which means at the moment all substrates have been converted into the most stable product). We can at this moment in time explain a non-enzymatic glycolysis but not (yet) a non-enzymatic gluconeogenesis (nor a non-enzymatic Calvin cycle, perhaps). Not ‘yet’ because, as said, the same laws of thermodynamics apply both to enzymatic and non-enzymatic reactions. If an enzymatic gluconeogenesis is possible, a non-enzymatic gluconeogenesis is thermodynamically possible as well. It is the question to find the reaction condition, that might very well be an unexpected one, and/or the required catalyst. This is not a trivial problem indeed, but one we hope to solve some day perhaps.
Which brings us, finally, to the present paper addressing non-enzymatic reactions that reflect reactivity as it occurs in the Krebs cycle (Keller et al. 2017). In contrast to glycolysis and the PPP which haven’t received much attention from the origin of life community, the Krebs cycle has received a lot of attention instead. The main motivation - I think at least initially - was that the cyclic structure of this metabolic pathway immediately implies a scenario which avoids the equilibrium problem, in addition to its dependency on iron-sulfur clusters that might resemble mineral surfaces. Furthermore, if the Krebs cycle would run in ‘reverse’ (meaning in the reductive directionality) it could fix carbon, and apparently, the Krebs cycle does so in some species (Buchanan and Arnon 1990). Nonetheless, already early systematic metagenome analysis have shown that the Krebs cycle is ‘complete’ in only a small subset of species, and hence it is unlikely that the full cycle was its evolutionary starting point (Huynen, Dandekar, and Bork 1999).
For us, the attractive point about the Krebs cycle was that its intermediates have been discovered in several non-enzymatic scenarios, like in the situation of carbonaceous meteorites by scientists from NASA (Cooper et al. 2011), or in non-enzymatic conditions that are prebiotically plausible, but not compatible with life and that depend on semiconductor catalysts not used by cells (Zhang and Martin 2006). Hence, the problem of a lack for substrates as currently for glycolysis and the PPP does not apply to the same extent to the Krebs cycle. Therefore, finding a biologically plausible (= life-compatible) condition that would enable a critical amount of Krebs cycle reactions would provide substantial proof for the hypothesis about a non-enzymatic origin of its network topology. The downside of our endeavor, however, was that some scientists had dismissed this possibility. Leslie Orgel, a leading figure in shaping the RNA world hypothesis, for instance, had called the quest for a universal catalyst for multiple Krebs cycle reactions an ‘appeal to magic’ (L. E. Orgel 2000). There is little motivation to conduct the experiment if you believe a splash of unicorn blood and dragon claw must be added to your reaction mix (as these are not listed in the Sigma catalogue and hence hard to obtain), and equally disheartening, you must also hope that your future reviewers have never read or memorized such statements: emotions play an important role if you attempt to publish in the origin of life field, especially if you’re (kind-of) young and not a chemist in the classic sense (you may want to glimpse into the blog or papers as referenced above).
Indeed, at high temperatures, no Krebs-cycle-like reactivity was observed, which shows that the enabling chemistry for the Krebs cycle is more complicated than the one behind glycolysis. This situation also did not change upon adding FeS as a simple mimetic for a mineral catalyst. However, my lab’s strengths lie in conducting large systematic screens using mass spectrometry (that is to say, our bread & butter) for my main research interest in Functional Metabolomics. We operate several mass spectrometers ourselves, and have methods optimized to process large and very large sample series (Mülleder et al. 2016). To address the Krebs cycle problem we combined, in essence, everything available in the lab which contained sulfur and iron, and tested all possible triple combinations (Fe + S + TCA metabolites) in a quantitative time series analysis. This screen included peroxydisulfate, which we happened to have in the lab owing to its use as a radical starter for the polymerisation of acrylamide gels in Western Blotting (which is a common technique in protein biochemistry). And we found an enabling condition: sulfate radicals that form from peroxydisulfate. So, in a way we were lucky to find the enabling reaction condition, even though we had approached the problem using a systematic screening experiment. Indeed, I doubt we would have ever specifically ordered peroxydisulfate in order to test its effect on Krebs cycle intermediates. So non-enzymatic Krebs cycle-like reactions enabled by a simple agent are not an ‘appeal to magic’ after all!
I don't want to go much further into detail here (please read the manuscript by Keller et al in the current issue of Nature Ecology and Evolution (doi: 10.1038/s41559-017-0083) if this Blog triggered your interest), but I would like to mention that these reactions are much more efficient than the ones enabling glycolysis, and that both non-enzymatic glycolytic and Krebs cycle-like reactions depend on just one key compound (Fe(II) for glycolysis, sulfate radicals for the TCA reactions). Both pathway chemistries hence could have emerged in simple, but distinct, prebiotic environments. How can they help to explain the origin of metabolism? We think that non-enzymatic chemical networks can serve as template to overcome the above-mentioned chicken-egg problem in imagining the origin of a metabolic pathway: if a chemical network exists, not all the enzymes that eventually form a metabolic pathway need to come into being at the same time to form a functional unit. A stepwise Darwinian scenario is enabled (Ralser 2014).
So what is missing, what are the limits of our study? First, we know little about the molecular reaction mechanisms that drive the non-enzymatic Krebs cycle-like reactions, other than that they depend on sulfate radicals. Some of the mechanisms seem to involve highly complicated, multistep reaction mechanisms. Solving these mechanisms in chemical, molecular detail, especially those that involve iron chemistry, exceed the possibilities of my biology-oriented laboratory. Hence, I hope these new reactions may stimulate the interest of one or more specialized organic chemists to work out chemical mechanisms (as it was the case for the reactions of a non-enzymatic glycolysis, for instance (Coggins and Powner 2016). Second, while the sulfate-radical enabled reactions form a Krebs-cycle like chemical network, this network is clearly not yet the complete biochemical ‘cycle’ as it operates within the modern cell. The C-C forming reaction as catalyzed by citrate synthase does not occur and we have not studied if we can couple the reactions to a co-enzyme-like chemistry, such as one involving NAD(P)H or acetyl-CoA, for instance. As stated, we do not think it is likely that the TCA cycle started as a full cycle in early evolution. Nonetheless, at some point the citrate synthase reaction and coenzyme coupling came into place. The evolutionary origin of these mechanisms is another, and important, puzzle to solve. Could it again be non-enzymatic reactions, or is the emergence of those a speciality of the enzymatic world, as Leslie Orgel once suggested (Leslie E. Orgel 2008)? I don’t know the answer - but it remains for me to repeat the mantra that is so easy to remember, yet at the same time so difficult for us biochemists to accept: the laws of thermodynamics that apply to non-enzymatic reactions are the same as the ones that apply to enzymatic reactions.
The paper in Nature Ecology & Evolution is available here: http://go.nature.com/2mVriLg
References
Coggins, Adam J., and Matthew W. Powner. 2016. “Prebiotic Synthesis of Phosphoenol Pyruvate by α-Phosphorylation-Controlled Triose Glycolysis.” Nature Chemistry, October. Nature Research. doi:10.1038/nchem.2624.
Keller, Markus A., Domen Kampjut, Stuart Harrison, and Markus Ralser. 2017. “Sulfate Radicals Enable a Non-Enzymatic Krebs Cycle Precursor.” Nature Ecology and Evolution in press. doi:10.1038/s41559-017-0083.
Luisi, Pier-Luigi. 2014. “Prebiotic Metabolic Networks?” Molecular Systems Biology 10 (4). EMBO Press. doi:10.1002/msb.20145351.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in